

Genome-wide discovery of somatic regulatory variants in diffuse large B-cell lymphoma
- PMID: 30275490
- PMCID: PMC6167379
- DOI: 10.1038/s41467-018-06354-3
Genome-wide discovery of somatic regulatory variants in diffuse large B-cell lymphoma
Abstract
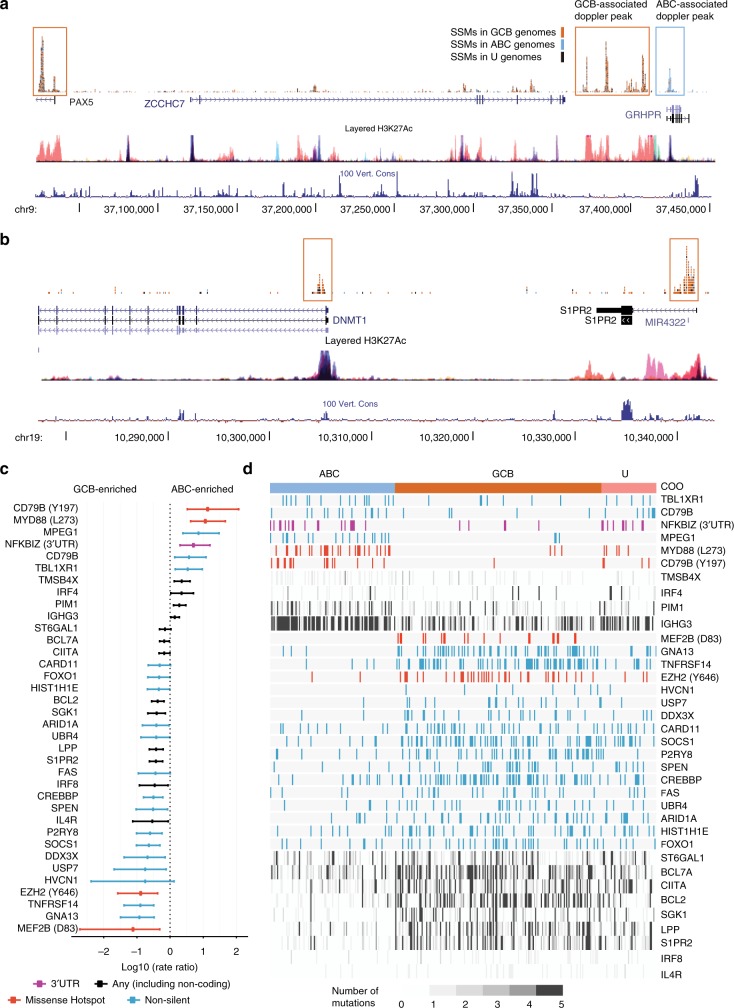
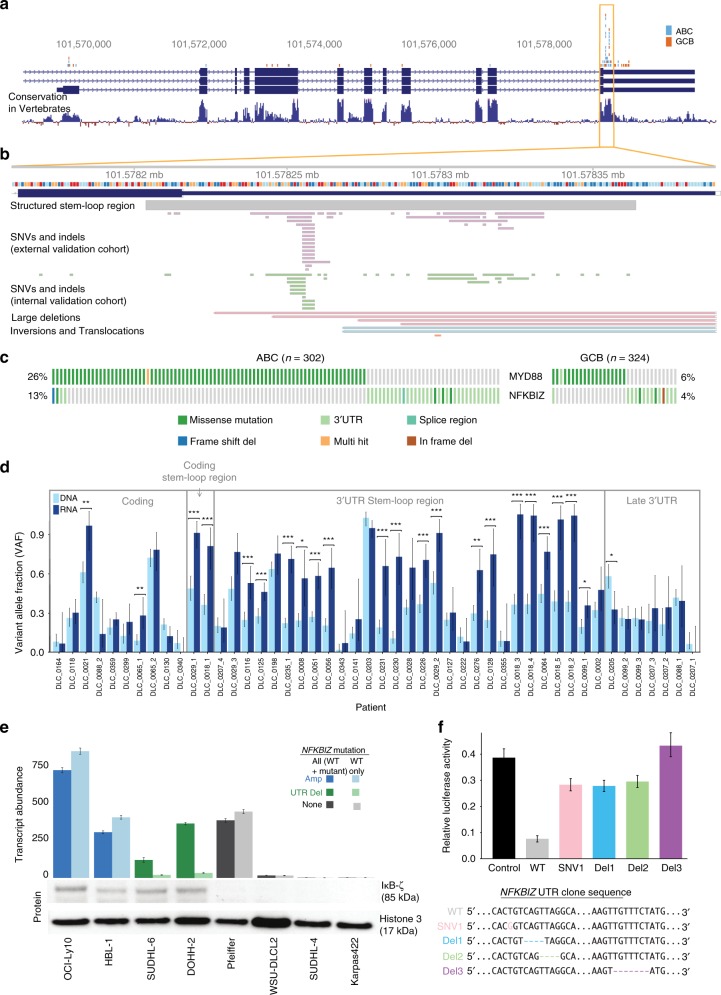
Diffuse large B-cell lymphoma (DLBCL) is an aggressive cancer originating from mature B-cells. Prognosis is strongly associated with molecular subgroup, although the driver mutations that distinguish the two main subgroups remain poorly defined. Through an integrative analysis of whole genomes, exomes, and transcriptomes, we have uncovered genes and non-coding loci that are commonly mutated in DLBCL. Our analysis has identified novel cis-regulatory sites, and implicates recurrent mutations in the 3' UTR of NFKBIZ as a novel mechanism of oncogene deregulation and NF-κB pathway activation in the activated B-cell (ABC) subgroup. Small amplifications associated with over-expression of FCGR2B (the Fcγ receptor protein IIB), primarily in the germinal centre B-cell (GCB) subgroup, correlate with poor patient outcomes suggestive of a novel oncogene. These results expand the list of subgroup driver mutations that may facilitate implementation of improved diagnostic assays and could offer new avenues for the development of targeted therapeutics.
Conflict of interest statement
The authors declare no competing interests.
Figures


References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
