

Predictors for a dementia gene mutation based on gene-panel next-generation sequencing of a large dementia referral series
- PMID: 30279455
- PMCID: PMC6330090
- DOI: 10.1038/s41380-018-0224-0
Predictors for a dementia gene mutation based on gene-panel next-generation sequencing of a large dementia referral series
Abstract
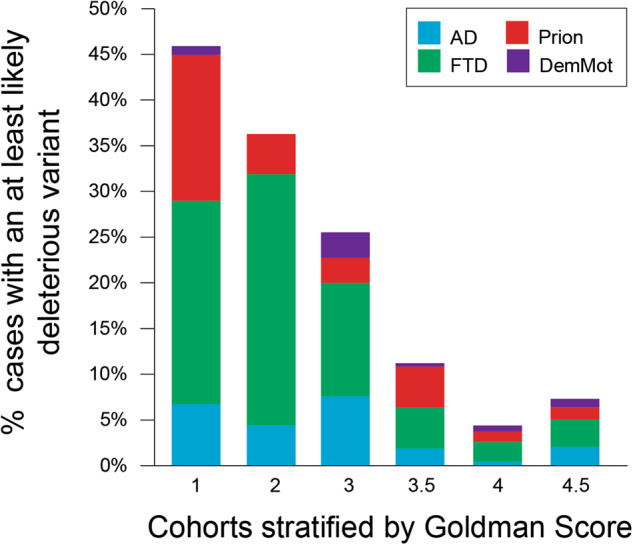
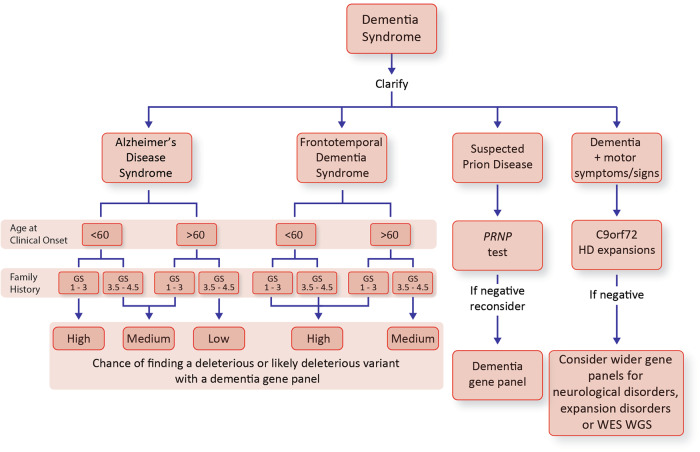
Next-generation genetic sequencing (NGS) technologies facilitate the screening of multiple genes linked to neurodegenerative dementia, but there are few reports about their use in clinical practice. Which patients would most profit from testing, and information on the likelihood of discovery of a causal variant in a clinical syndrome, are conspicuously absent from the literature, mostly for a lack of large-scale studies. We applied a validated NGS dementia panel to 3241 patients with dementia and healthy aged controls; 13,152 variants were classified by likelihood of pathogenicity. We identified 354 deleterious variants (DV, 12.6% of patients); 39 were novel DVs. Age at clinical onset, clinical syndrome and family history each strongly predict the likelihood of finding a DV, but healthcare setting and gender did not. DVs were frequently found in genes not usually associated with the clinical syndrome. Patients recruited from primary referral centres were compared with those seen at higher-level research centres and a national clinical neurogenetic laboratory; rates of discovery were comparable, making selection bias unlikely and the results generalisable to clinical practice. We estimated penetrance of DVs using large-scale online genomic population databases and found 71 with evidence of reduced penetrance. Two DVs in the same patient were found more frequently than expected. These data should provide a basis for more informed counselling and clinical decision making.
Conflict of interest statement
Through UCL Consultants Ltd, a wholly owned subsidiary of University College London, EJW has served on scientific advisory boards for Novartis, F Hoffmann-La Roche, Ionis, Shire, Wave Life Sciences, PTC Therapeutics and Mitoconix.
Prof Collinge is a director and shareholder of D-Gen Limited (London), an academic spinout company working in the field of prion disease diagnosis, decontamination, and therapeutics.
None of the other authors report any conflict of interest.
Figures
References
Publication types
MeSH terms
Grants and funding
- MR/K013041/1/MRC_/Medical Research Council/United Kingdom
- MC_UU_00024/1/MRC_/Medical Research Council/United Kingdom
- G0300429/MRC_/Medical Research Council/United Kingdom
- MC_U105597119/MRC_/Medical Research Council/United Kingdom
- G0902227/MRC_/Medical Research Council/United Kingdom
- MR/L501517/1/MRC_/Medical Research Council/United Kingdom
- MR/L023784/1/MRC_/Medical Research Council/United Kingdom
- MR/L012936/1/MRC_/Medical Research Council/United Kingdom
- MR/M009076/1/MRC_/Medical Research Council/United Kingdom
- G0600237/MRC_/Medical Research Council/United Kingdom
- MR/M008592/1/MRC_/Medical Research Council/United Kingdom
- 103838/WT_/Wellcome Trust/United Kingdom
- MR/L02053X/1/MRC_/Medical Research Council/United Kingdom
- 200181/Z/15/Z/WT_/Wellcome Trust/United Kingdom
- MR/J009482/1/MRC_/Medical Research Council/United Kingdom
- MR/M023664/1/MRC_/Medical Research Council/United Kingdom
- BRC149/NS/MH/DH_/Department of Health/United Kingdom
- MR/L023784/2/MRC_/Medical Research Council/United Kingdom
- MC_UU_00005/12/MRC_/Medical Research Council/United Kingdom
- MR/M008525/1/MRC_/Medical Research Council/United Kingdom
- G9810900/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Medical
