

Functional equivalence of genome sequencing analysis pipelines enables harmonized variant calling across human genetics projects
- PMID: 30279509
- PMCID: PMC6168605
- DOI: 10.1038/s41467-018-06159-4
Functional equivalence of genome sequencing analysis pipelines enables harmonized variant calling across human genetics projects
Abstract
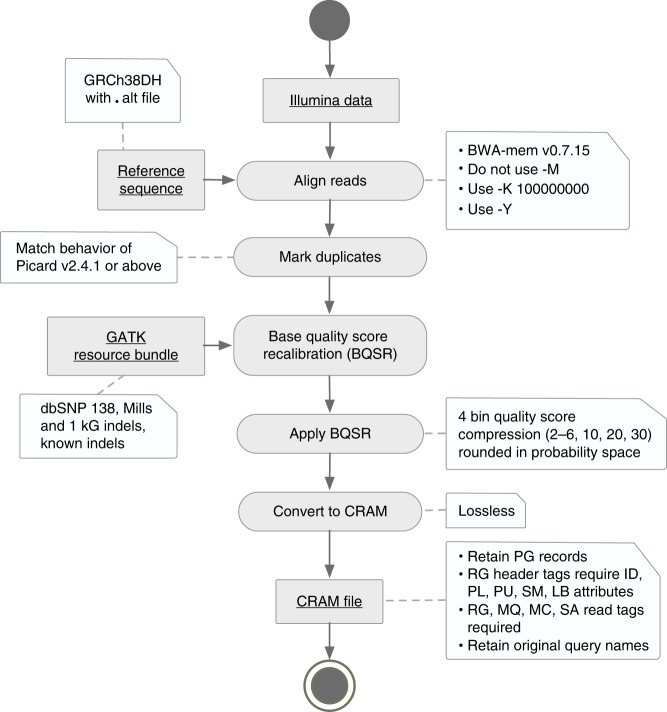
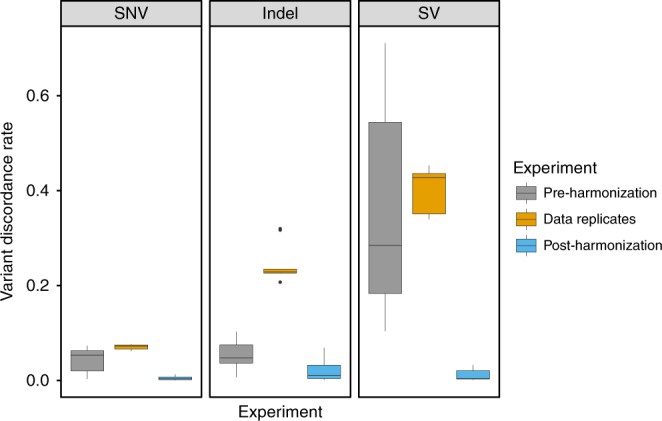
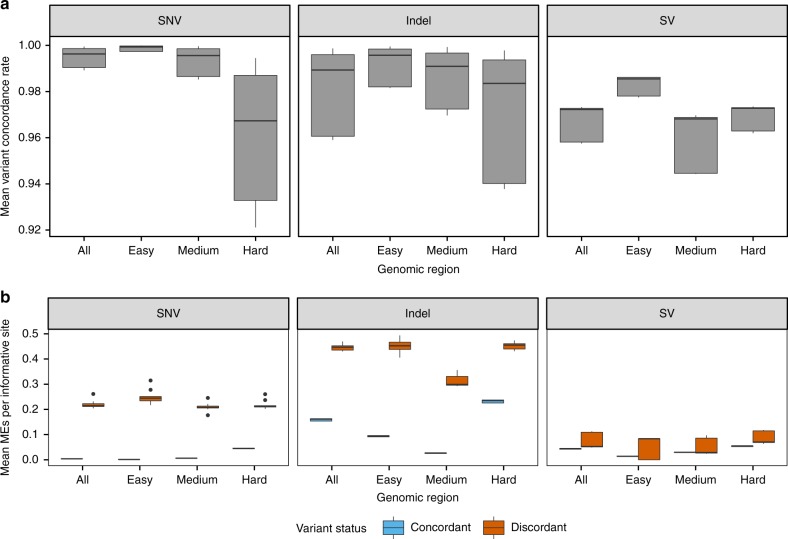
Hundreds of thousands of human whole genome sequencing (WGS) datasets will be generated over the next few years. These data are more valuable in aggregate: joint analysis of genomes from many sources increases sample size and statistical power. A central challenge for joint analysis is that different WGS data processing pipelines cause substantial differences in variant calling in combined datasets, necessitating computationally expensive reprocessing. This approach is no longer tenable given the scale of current studies and data volumes. Here, we define WGS data processing standards that allow different groups to produce functionally equivalent (FE) results, yet still innovate on data processing pipelines. We present initial FE pipelines developed at five genome centers and show that they yield similar variant calling results and produce significantly less variability than sequencing replicates. This work alleviates a key technical bottleneck for genome aggregation and helps lay the foundation for community-wide human genetics studies.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Caulfield, M. et al. The 100,000 Genomes Project Protocol. figshare 10.6084/m9.figshare.4530893.v2 (2017).
-
- Alliance Aviesan. Genomic Medicine France 2025 (Aviesan, 2017).
-
- Felsenfeld, A. Centers for Common Disease Genomics. National Human Genome Research Institute, https://www.genome.gov/27563570 (2016).
Publication types
MeSH terms
Grants and funding
- UM1 HG008853/U.S. Department of Health & Human Services | NIH | National Human Genome Research Institute (NHGRI)/International
- UM1 HG008901/U.S. Department of Health & Human Services | NIH | National Human Genome Research Institute (NHGRI)/International
- UM1 HG008895/HG/NHGRI NIH HHS/United States
- UM1 HG008895/U.S. Department of Health & Human Services | NIH | National Heart, Lung, and Blood Institute (NHLBI)/International
- R01 HG002818/HG/NHGRI NIH HHS/United States
- U24 HG008956/U.S. Department of Health & Human Services | NIH | National Human Genome Research Institute (NHGRI)/International
- UM1 HG008901/HG/NHGRI NIH HHS/United States
- U01 HG00908/U.S. Department of Health & Human Services | NIH | National Human Genome Research Institute (NHGRI)/International
- UM1 HG008898/U.S. Department of Health & Human Services | NIH | National Human Genome Research Institute (NHGRI)/International
- UM1 HG008853/HG/NHGRI NIH HHS/United States
- 3R01HL-117626-02S1/U.S. Department of Health & Human Services | NIH | National Heart, Lung, and Blood Institute (NHLBI)/International
- R01 MH107649/MH/NIMH NIH HHS/United States
- U01 HL137182/HL/NHLBI NIH HHS/United States
- UM1 HG008895/U.S. Department of Health & Human Services | NIH | National Human Genome Research Institute (NHGRI)/International
- R21 HL133758/HL/NHLBI NIH HHS/United States
- U01 HG009088/HG/NHGRI NIH HHS/United States
- UM1 HG008898/HG/NHGRI NIH HHS/United States
- R01 HL117626/HL/NHLBI NIH HHS/United States
- U24 HG008956/HG/NHGRI NIH HHS/United States
- R01 MH107649/U.S. Department of Health & Human Services | NIH | National Institute of Mental Health (NIMH)/International
- 4 R01 HL117626-04/U.S. Department of Health & Human Services | NIH | National Heart, Lung, and Blood Institute (NHLBI)/International
LinkOut - more resources
Full Text Sources
Other Literature Sources
