

Pepducin-mediated cardioprotection via β-arrestin-biased β2-adrenergic receptor-specific signaling
- PMID: 30279730
- PMCID: PMC6160776
- DOI: 10.7150/thno.26619
Pepducin-mediated cardioprotection via β-arrestin-biased β2-adrenergic receptor-specific signaling
Abstract
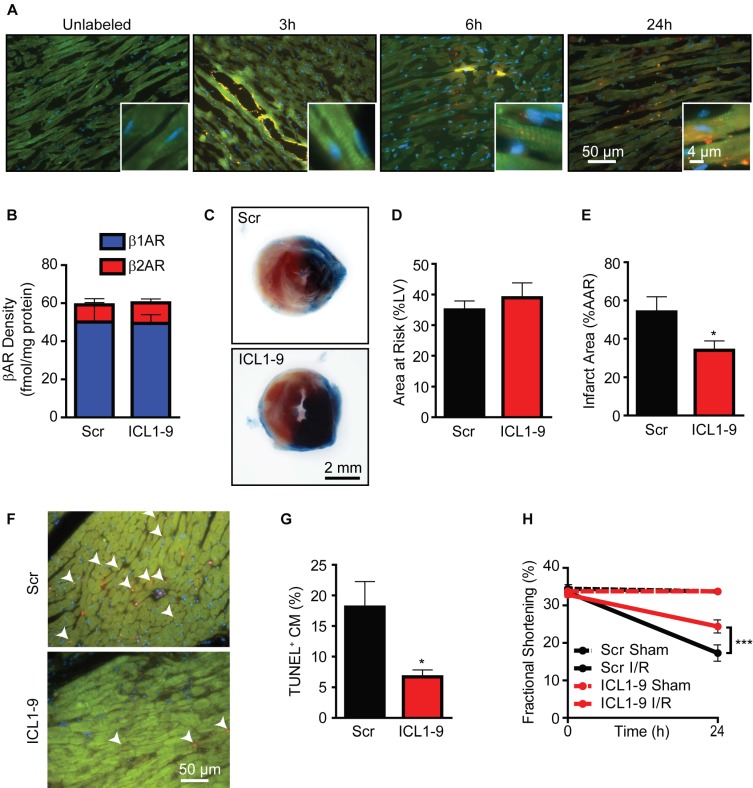
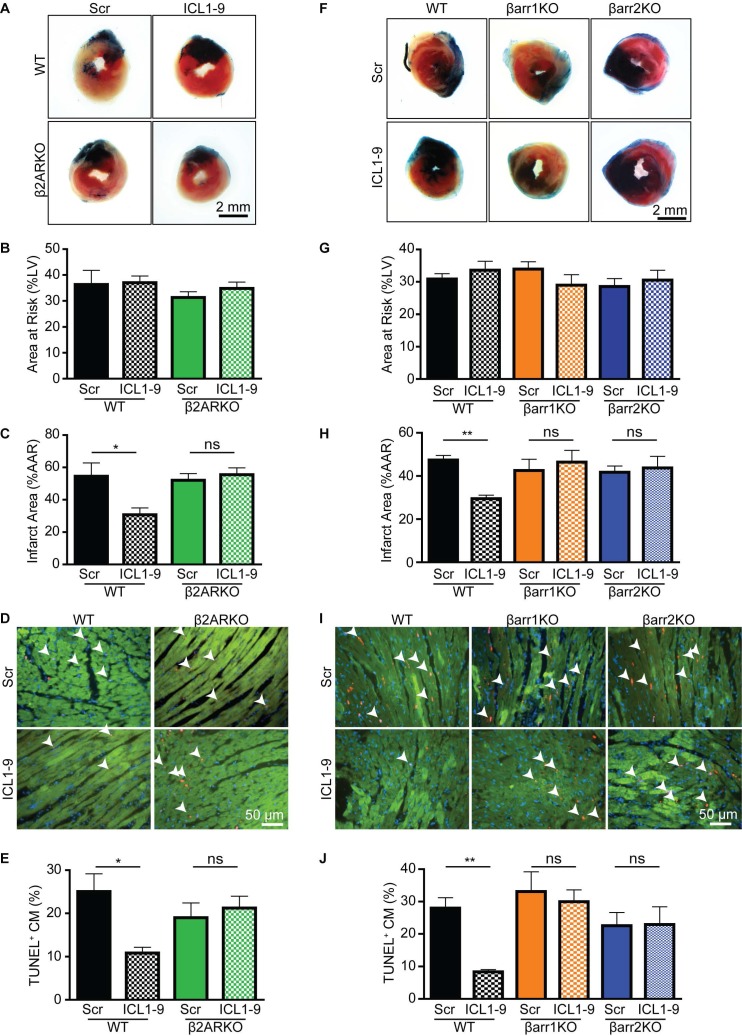
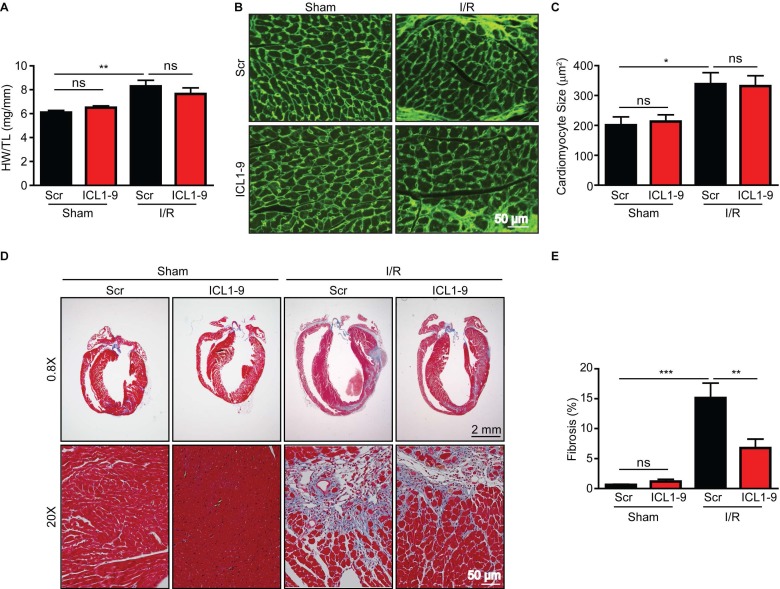
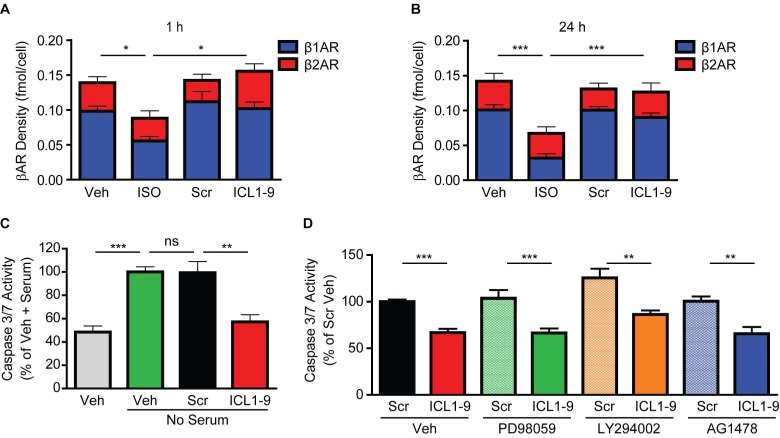
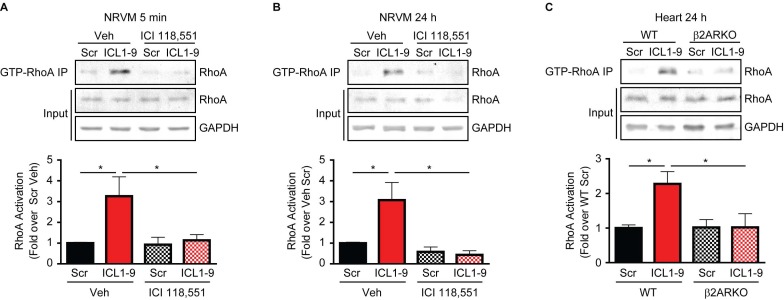
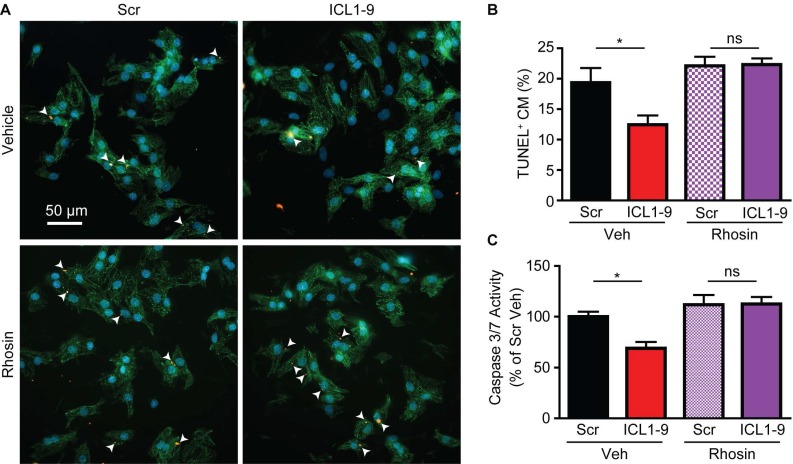
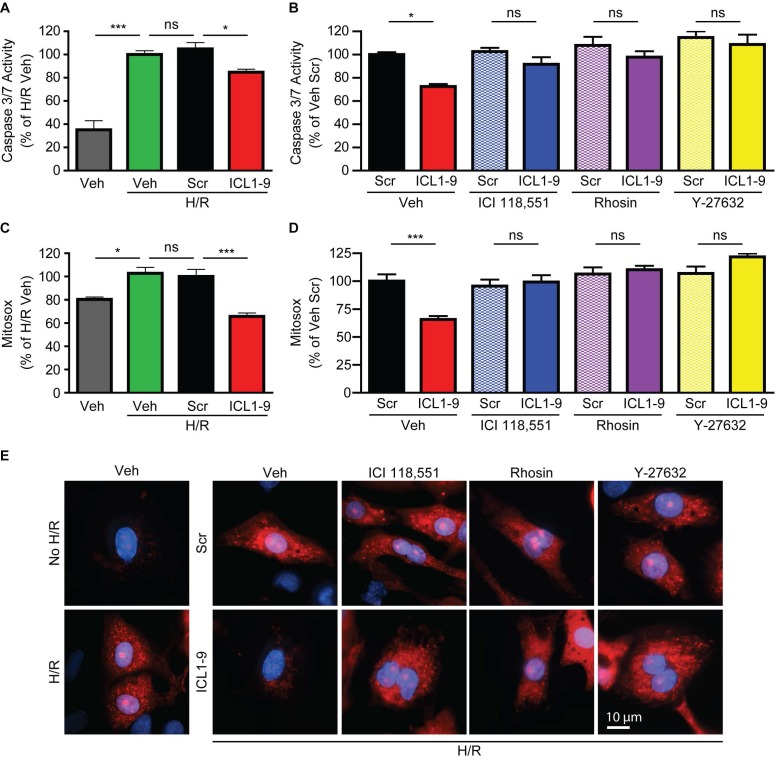
Reperfusion as a therapeutic intervention for acute myocardial infarction-induced cardiac injury itself induces further cardiomyocyte death. β-arrestin (βarr)-biased β-adrenergic receptor (βAR) activation promotes survival signaling responses in vitro; thus, we hypothesize that this pathway can mitigate cardiomyocyte death at the time of reperfusion to better preserve function. However, a lack of efficacious βarr-biased orthosteric small molecules has prevented investigation into whether this pathway relays protection against ischemic injury in vivo. We recently demonstrated that the pepducin ICL1-9, a small lipidated peptide fragment designed from the first intracellular loop of β2AR, allosterically engaged pro-survival signaling cascades in a βarr-dependent manner in vitro. Thus, in this study we tested whether ICL1-9 relays cardioprotection against ischemia/reperfusion (I/R)-induced injury in vivo. Methods: Wild-type (WT) C57BL/6, β2AR knockout (KO), βarr1KO and βarr2KO mice received intracardiac injections of either ICL1-9 or a scrambled control pepducin (Scr) at the time of ischemia (30 min) followed by reperfusion for either 24 h, to assess infarct size and cardiomyocyte death, or 4 weeks, to monitor the impact of ICL1-9 on long-term cardiac structure and function. Neonatal rat ventricular myocytes (NRVM) were used to assess the impact of ICL1-9 versus Scr pepducin on cardiomyocyte survival and mitochondrial superoxide formation in response to either serum deprivation or hypoxia/reoxygenation (H/R) in vitro and to investigate the associated mechanism(s). Results: Intramyocardial injection of ICL1-9 at the time of I/R reduced infarct size, cardiomyocyte death and improved cardiac function in a β2AR- and βarr-dependent manner, which led to improved contractile function early and less fibrotic remodeling over time. Mechanistically, ICL1-9 attenuated mitochondrial superoxide production and promoted cardiomyocyte survival in a RhoA/ROCK-dependent manner. RhoA activation could be detected in cardiomyocytes and whole heart up to 24 h post-treatment, demonstrating the stability of ICL1-9 effects on βarr-dependent β2AR signaling. Conclusion: Pepducin-based allosteric modulation of βarr-dependent β2AR signaling represents a novel therapeutic approach to reduce reperfusion-induced cardiac injury and relay long-term cardiac remodeling benefits.
Keywords: Pepducin; cardiac ischemia/reperfusion; cardiomyocyte; β-arrestin; β2-adrenergic receptor.
Conflict of interest statement
Competing Interests: The authors have declared that no competing interest exists.
Figures
References
-
- Jneid H, Addison D, Bhatt DL, Fonarow GC, Gokak S, Grady KL. et al. 2017 AHA/ACC clinical performance and quality measures for adults with ST-elevation and non-ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Performance Measures. J Am Coll Cardiol. 2017;70:2048–90. - PubMed
-
- Grisanti LA, Talarico JA, Carter RL, Yu JE, Repas AA, Radcliffe SW. et al. beta-Adrenergic receptor-mediated transactivation of epidermal growth factor receptor decreases cardiomyocyte apoptosis through differential subcellular activation of ERK1/2 and Akt. J Mol Cell Cardiol. 2014;72:39–51. - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
