

Blood-Brain Barrier: From Physiology to Disease and Back
- PMID: 30280653
- PMCID: PMC6335099
- DOI: 10.1152/physrev.00050.2017
Blood-Brain Barrier: From Physiology to Disease and Back
Abstract
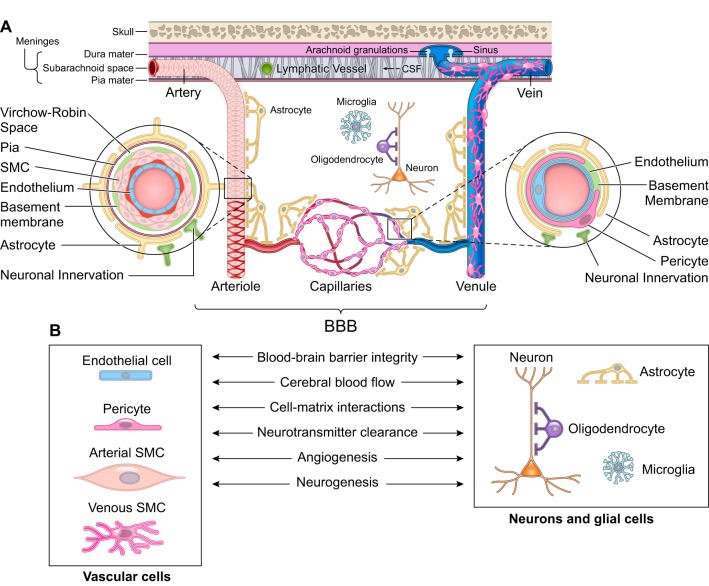
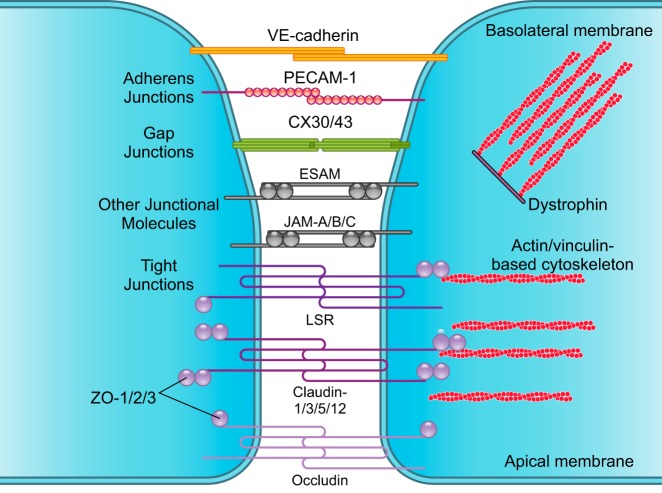
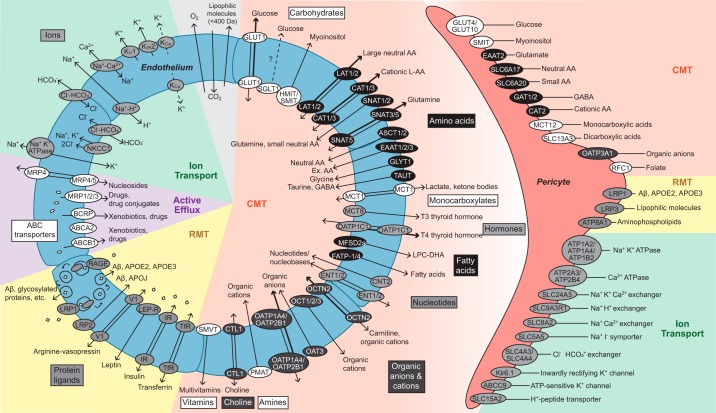
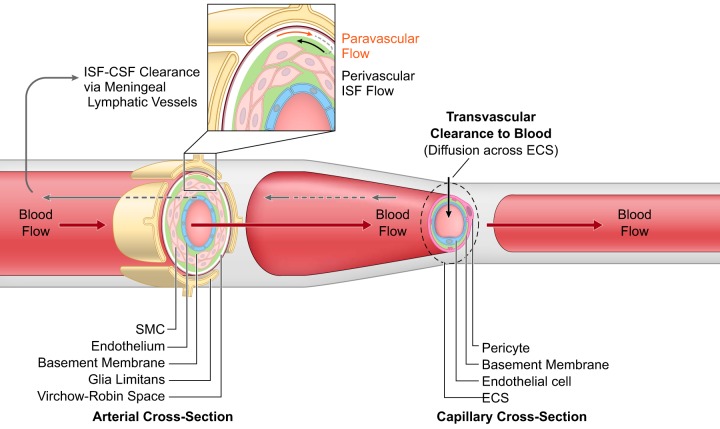
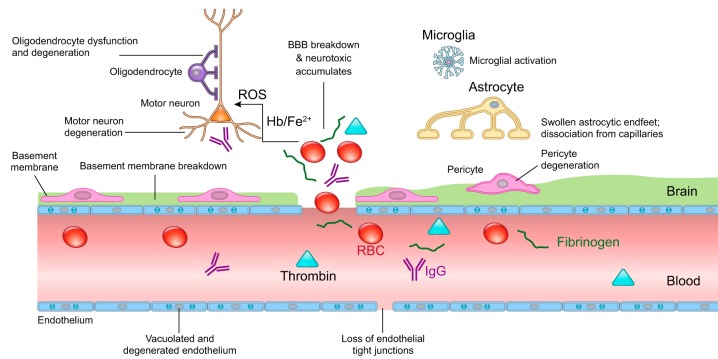
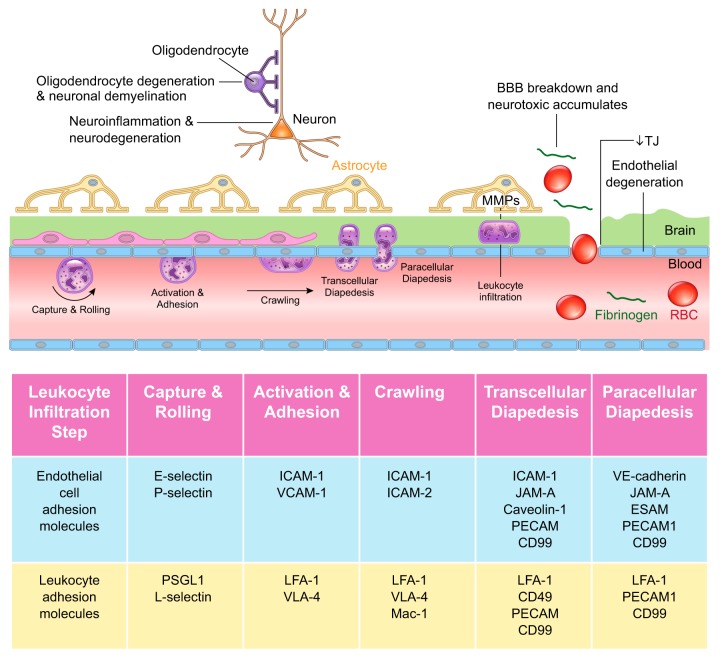
The blood-brain barrier (BBB) prevents neurotoxic plasma components, blood cells, and pathogens from entering the brain. At the same time, the BBB regulates transport of molecules into and out of the central nervous system (CNS), which maintains tightly controlled chemical composition of the neuronal milieu that is required for proper neuronal functioning. In this review, we first examine molecular and cellular mechanisms underlying the establishment of the BBB. Then, we focus on BBB transport physiology, endothelial and pericyte transporters, and perivascular and paravascular transport. Next, we discuss rare human monogenic neurological disorders with the primary genetic defect in BBB-associated cells demonstrating the link between BBB breakdown and neurodegeneration. Then, we review the effects of genes underlying inheritance and/or increased susceptibility for Alzheimer's disease (AD), Parkinson's disease (PD), Huntington's disease, and amyotrophic lateral sclerosis (ALS) on BBB in relation to other pathologies and neurological deficits. We next examine how BBB dysfunction relates to neurological deficits and other pathologies in the majority of sporadic AD, PD, and ALS cases, multiple sclerosis, other neurodegenerative disorders, and acute CNS disorders such as stroke, traumatic brain injury, spinal cord injury, and epilepsy. Lastly, we discuss BBB-based therapeutic opportunities. We conclude with lessons learned and future directions, with emphasis on technological advances to investigate the BBB functions in the living human brain, and at the molecular and cellular level, and address key unanswered questions.
Figures
Comment in
-
A definitive guide to the blood-brain barrier.Am J Physiol Regul Integr Comp Physiol. 2019 Jul 1;317(1):R14. doi: 10.1152/ajpregu.00073.2019. Epub 2019 Apr 3. Am J Physiol Regul Integr Comp Physiol. 2019. PMID: 30943048 Free PMC article. No abstract available.
References
-
- Abadier M, Haghayegh Jahromi N, Cardoso Alves L, Boscacci R, Vestweber D, Barnum S, Deutsch U, Engelhardt B, Lyck R. Cell surface levels of endothelial ICAM-1 influence the transcellular or paracellular T-cell diapedesis across the blood-brain barrier. Eur J Immunol 45: 1043–1058, 2015. doi: 10.1002/eji.201445125. - DOI - PubMed
-
- Afonso PV, Ozden S, Cumont M-C, Seilhean D, Cartier L, Rezaie P, Mason S, Lambert S, Huerre M, Gessain A, Couraud P-O, Pique C, Ceccaldi P-E, Romero IA. Alteration of blood-brain barrier integrity by retroviral infection. PLoS Pathog 4: e1000205, 2008. doi: 10.1371/journal.ppat.1000205. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous