

Cmah-dystrophin deficient mdx mice display an accelerated cardiac phenotype that is improved following peptide-PMO exon skipping treatment
- PMID: 30281092
- PMCID: PMC6337703
- DOI: 10.1093/hmg/ddy346
Cmah-dystrophin deficient mdx mice display an accelerated cardiac phenotype that is improved following peptide-PMO exon skipping treatment
Abstract
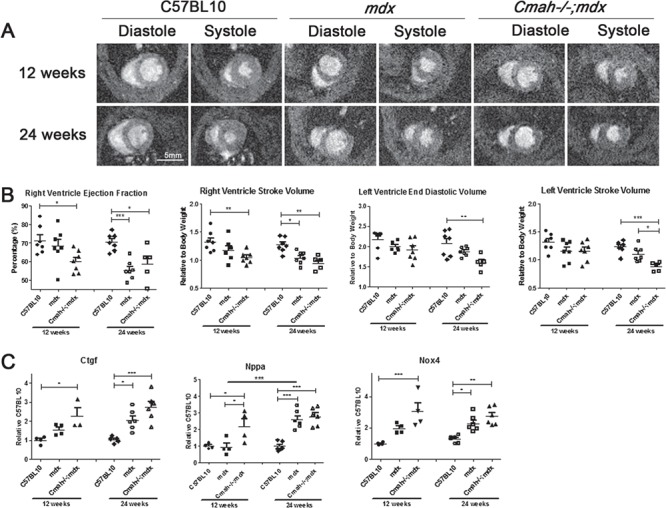
Duchenne muscular dystrophy (DMD) is caused by loss of dystrophin protein, leading to progressive muscle weakness and premature death due to respiratory and/or cardiac complications. Cardiac involvement is characterized by progressive dilated cardiomyopathy, decreased fractional shortening and metabolic dysfunction involving reduced metabolism of fatty acids-the major cardiac metabolic substrate. Several mouse models have been developed to study molecular and pathological consequences of dystrophin deficiency, but do not recapitulate all aspects of human disease pathology and exhibit a mild cardiac phenotype. Here we demonstrate that Cmah (cytidine monophosphate-sialic acid hydroxylase)-deficient mdx mice (Cmah-/-;mdx) have an accelerated cardiac phenotype compared to the established mdx model. Cmah-/-;mdx mice display earlier functional deterioration, specifically a reduction in right ventricle (RV) ejection fraction and stroke volume (SV) at 12 weeks of age and decreased left ventricle diastolic volume with subsequent reduced SV compared to mdx mice by 24 weeks. They further show earlier elevation of cardiac damage markers for fibrosis (Ctgf), oxidative damage (Nox4) and haemodynamic load (Nppa). Cardiac metabolic substrate requirement was assessed using hyperpolarized magnetic resonance spectroscopy indicating increased in vivo glycolytic flux in Cmah-/-;mdx mice. Early upregulation of mitochondrial genes (Ucp3 and Cpt1) and downregulation of key glycolytic genes (Pdk1, Pdk4, Ppara), also denote disturbed cardiac metabolism and shift towards glucose utilization in Cmah-/-;mdx mice. Moreover, we show long-term treatment with peptide-conjugated exon skipping antisense oligonucleotides (20-week regimen), resulted in 20% cardiac dystrophin protein restoration and significantly improved RV cardiac function. Therefore, Cmah-/-;mdx mice represent an appropriate model for evaluating cardiac benefit of novel DMD therapeutics.
Figures




References
-
- Vry J., Gramsch K., Rodger S., Thompson R., Steffensen B.F., Rahbek J., Doerken S., Tassoni A., Beytia M.L., Guergueltcheva V. et al. (2016) European cross-sectional survey of current care practices for Duchenne muscular dystrophy reveals regional and age-dependent differences. J. Neuromuscul. Dis., 3, 517–527. - PMC - PubMed
-
- Eagle M., Baudouin S.V., Chandler C., Giddings D.R., Bullock R. and Bushby K. (2002) Survival in Duchenne muscular dystrophy: improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromuscul. Disord., 12, 926–929. - PubMed
-
- Nigro G., Comi L.I., Politano L. and Bain R.J. (1990) The incidence and evolution of cardiomyopathy in Duchenne muscular dystrophy. Int. J. Cardiol., 26, 271–277. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- RG/11/9/28921/BHF_/British Heart Foundation/United Kingdom
- FS/10/002/28078/BHF_/British Heart Foundation/United Kingdom
- PG/14/2/30595/BHF_/British Heart Foundation/United Kingdom
- MR/N024850/1/MRC_/Medical Research Council/United Kingdom
- RG/11/9/28921 /BHF_/British Heart Foundation/United Kingdom
- MR/R025312/1/MRC_/Medical Research Council/United Kingdom
- FS/10/002/28078 /BHF_/British Heart Foundation/United Kingdom
- FS/19/18/34252/BHF_/British Heart Foundation/United Kingdom
- FS/14/17/30634/BHF_/British Heart Foundation/United Kingdom
- G0900887/MRC_/Medical Research Council/United Kingdom
- G0601490/MRC_/Medical Research Council/United Kingdom
- MC_U105178803/MRC_/Medical Research Council/United Kingdom
- MR/L013142/1/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
