

Spinocerebellar ataxia: an update
- PMID: 30284037
- PMCID: PMC6373366
- DOI: 10.1007/s00415-018-9076-4
Spinocerebellar ataxia: an update
Abstract
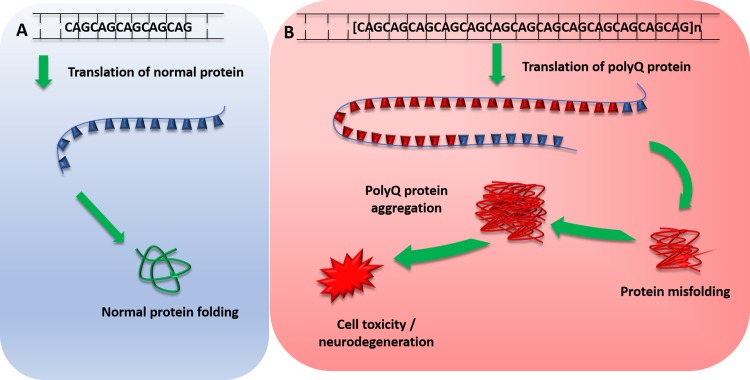
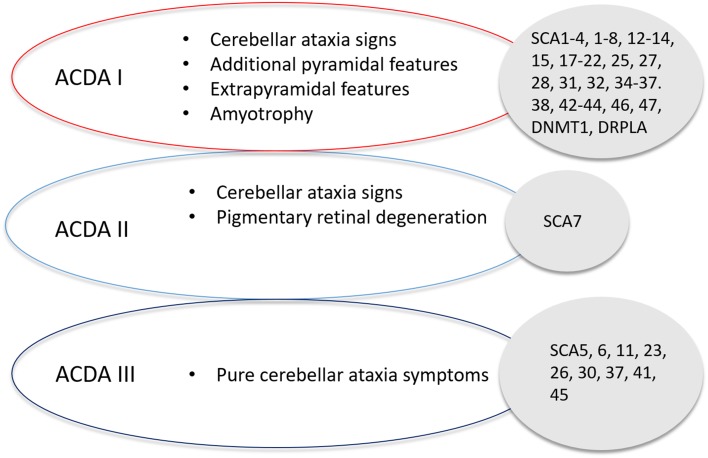
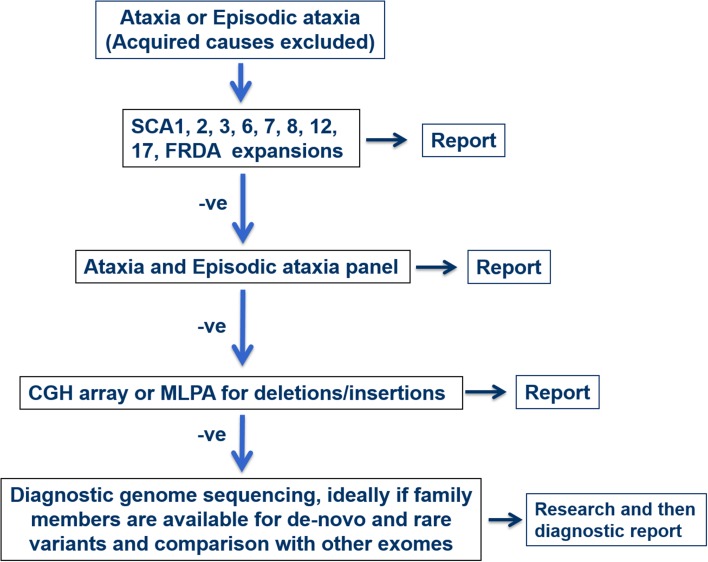
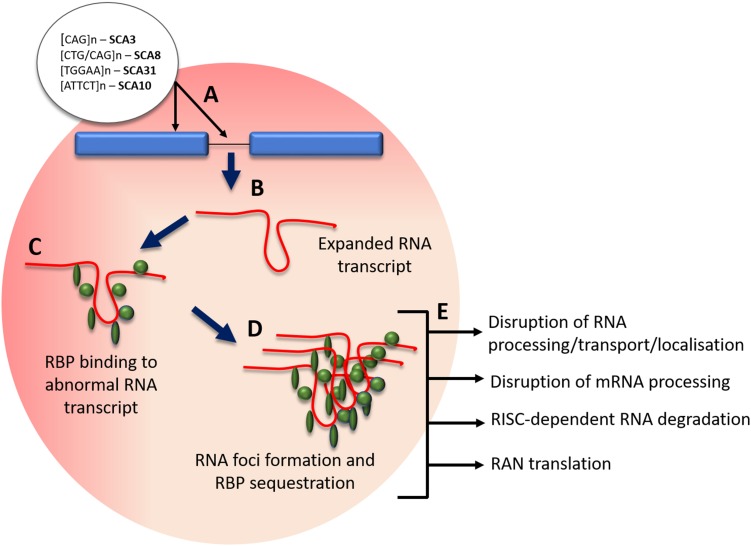
Spinocerebellar ataxia (SCA) is a heterogeneous group of neurodegenerative ataxic disorders with autosomal dominant inheritance. We aim to provide an update on the recent clinical and scientific progresses in SCA where numerous novel genes have been identified with next-generation sequencing techniques. The main disease mechanisms of these SCAs include toxic RNA gain-of-function, mitochondrial dysfunction, channelopathies, autophagy and transcription dysregulation. Recent studies have also demonstrated the importance of DNA repair pathways in modifying SCA with CAG expansions. In addition, we summarise the latest technological advances in detecting known and novel repeat expansion in SCA. Finally, we discuss the roles of antisense oligonucleotides and RNA-based therapy as potential treatments.
Keywords: Molecular diagnosis; Next-generation sequencing; Spinocerebellar ataxia.
Conflict of interest statement
This article does not contain any studies with human participants performed by any of the authors.
Figures
References
-
- Ruano L, Melo C, Silva MC, Coutinho P. The global epidemiology of hereditary ataxia and spastic paraplegia: a systematic review of prevalence studies. Neuroepidemiology. 2014;42(3):174–183. - PubMed
-
- de Castilhos RM, Furtado GV, Gheno TC, Schaeffer P, Russo A, Barsottini O, et al. Spinocerebellar ataxias in Brazil–frequencies and modulating effects of related genes. Cerebellum. 2014;13(1):17–28. - PubMed
-
- Coutinho P, Ruano L, Loureiro JL, Cruz VT, Barros J, Tuna A, et al. Hereditary ataxia and spastic paraplegia in Portugal: a population-based prevalence study. JAMA Neurol. 2013;70(6):746–755. - PubMed
-
- Zaltzman R, Sharony R, Klein C, Gordon CR. Spinocerebellar ataxia type 3 in Israel: phenotype and genotype of a Jew Yemenite subpopulation. J Neurol. 2016;263(11):2207–2214. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
