

Knockout of ClC-2 reveals critical functions of adherens junctions in colonic homeostasis and tumorigenicity
- PMID: 30285466
- PMCID: PMC6336945
- DOI: 10.1152/ajpgi.00087.2018
Knockout of ClC-2 reveals critical functions of adherens junctions in colonic homeostasis and tumorigenicity
Abstract
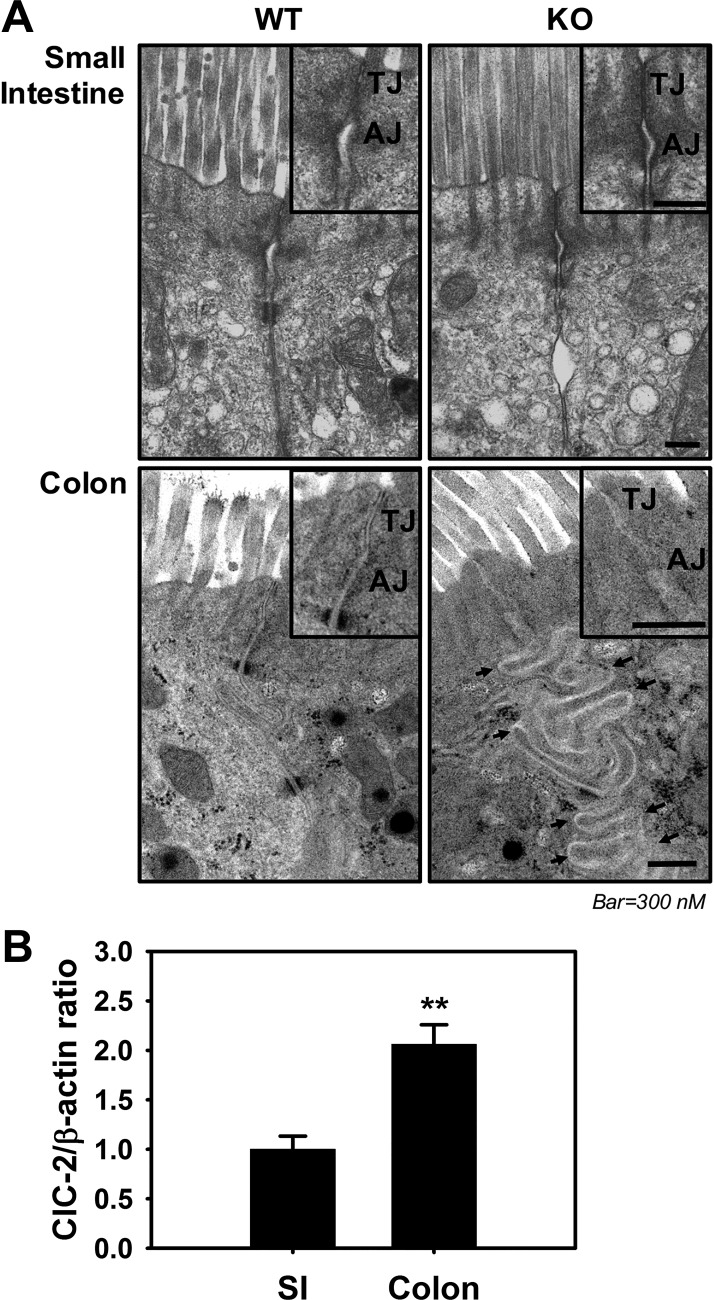
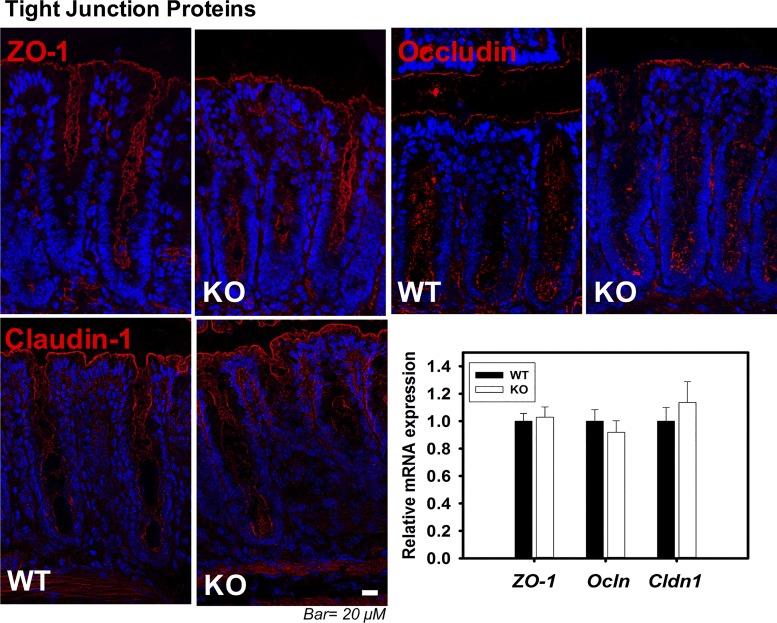
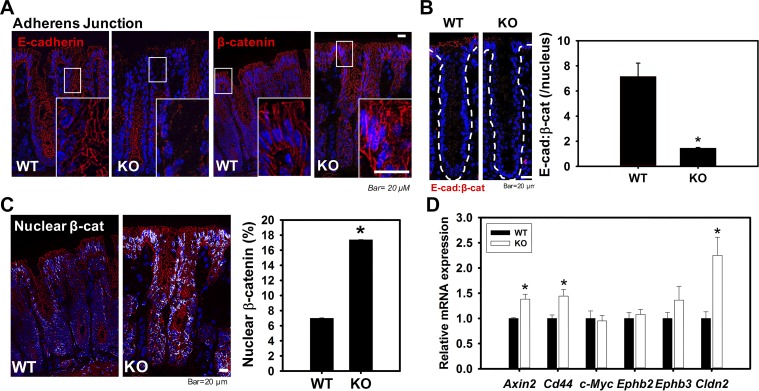
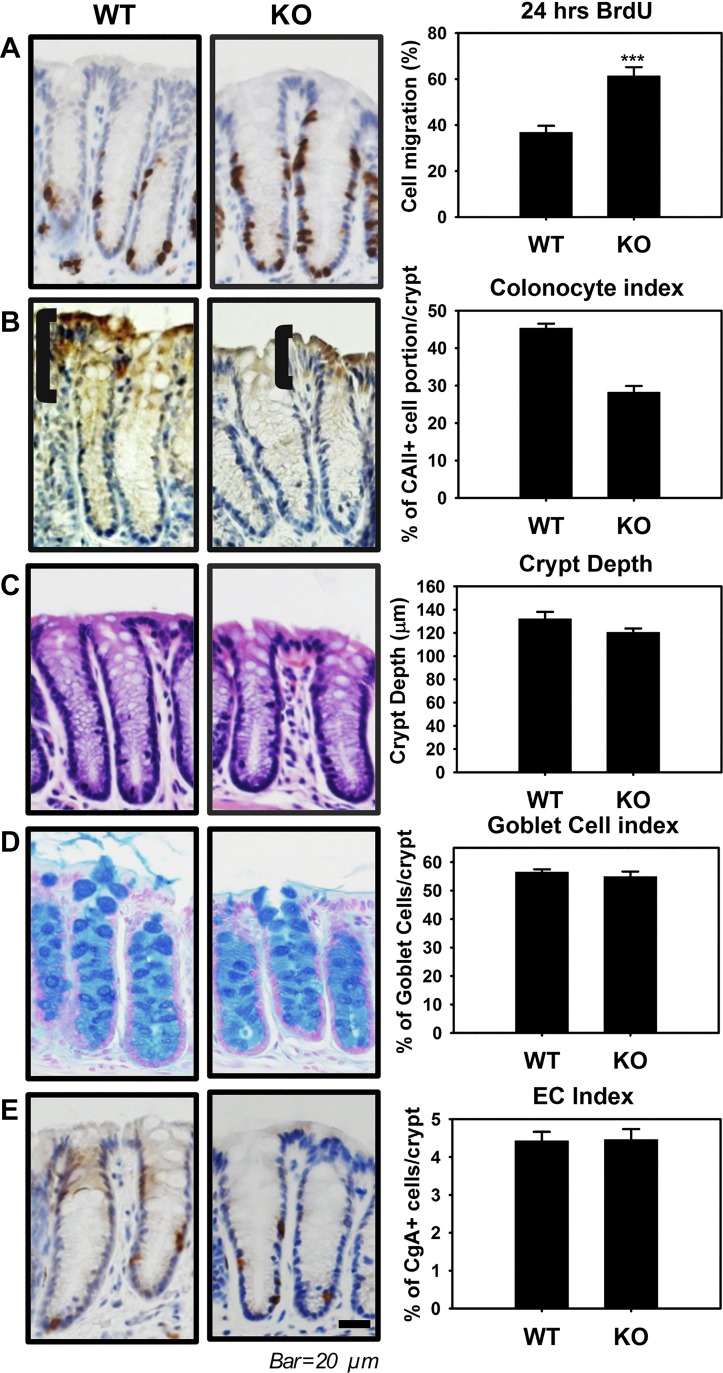
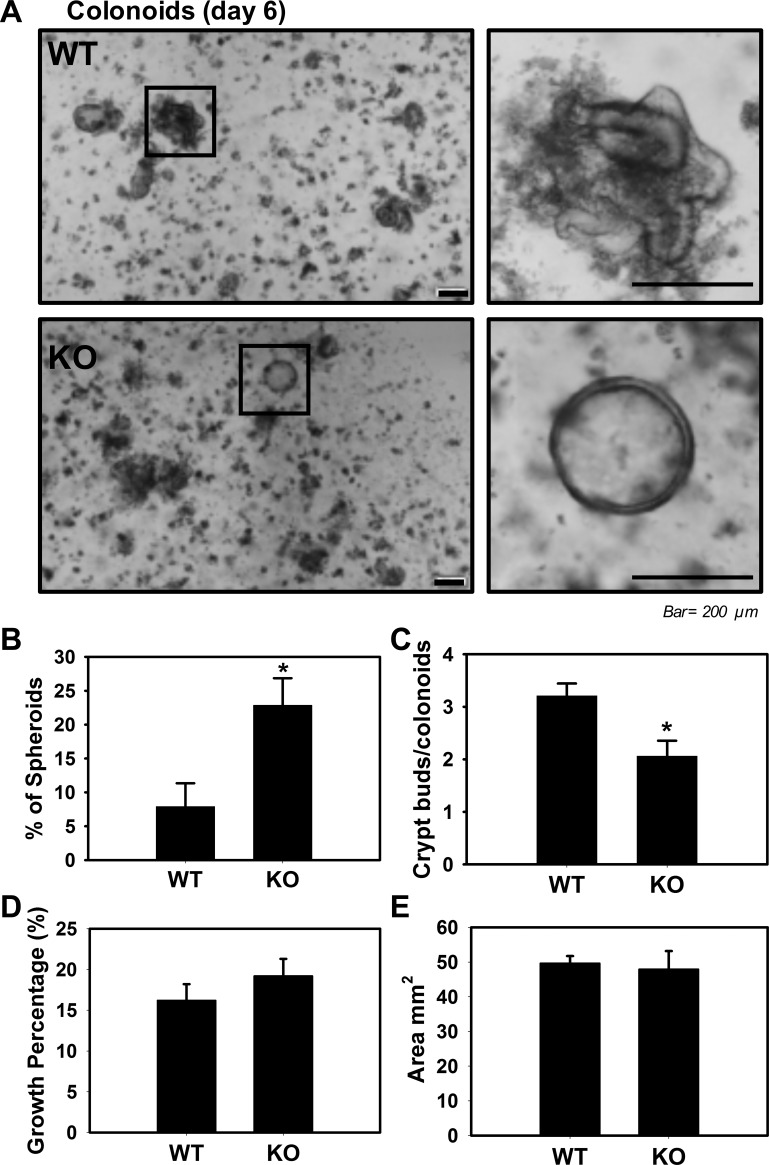
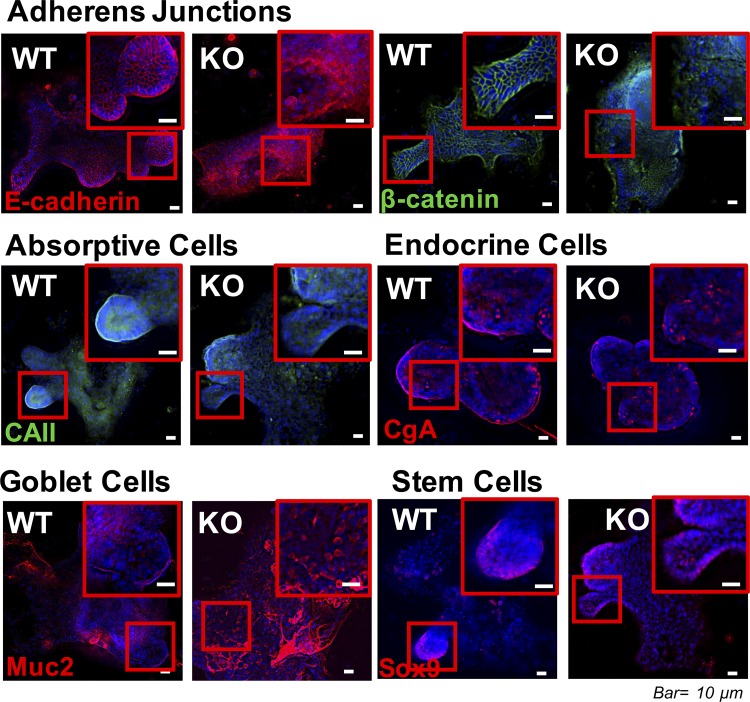
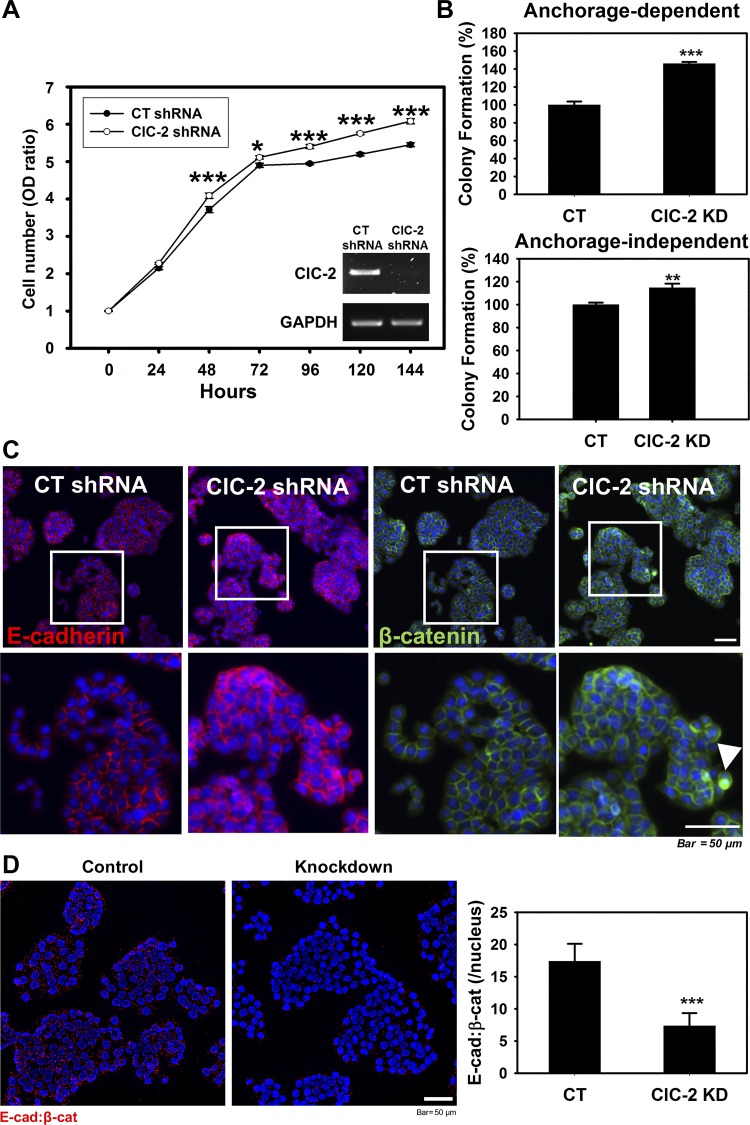
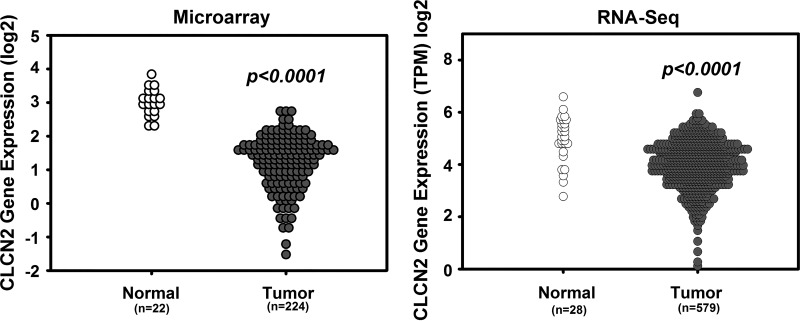
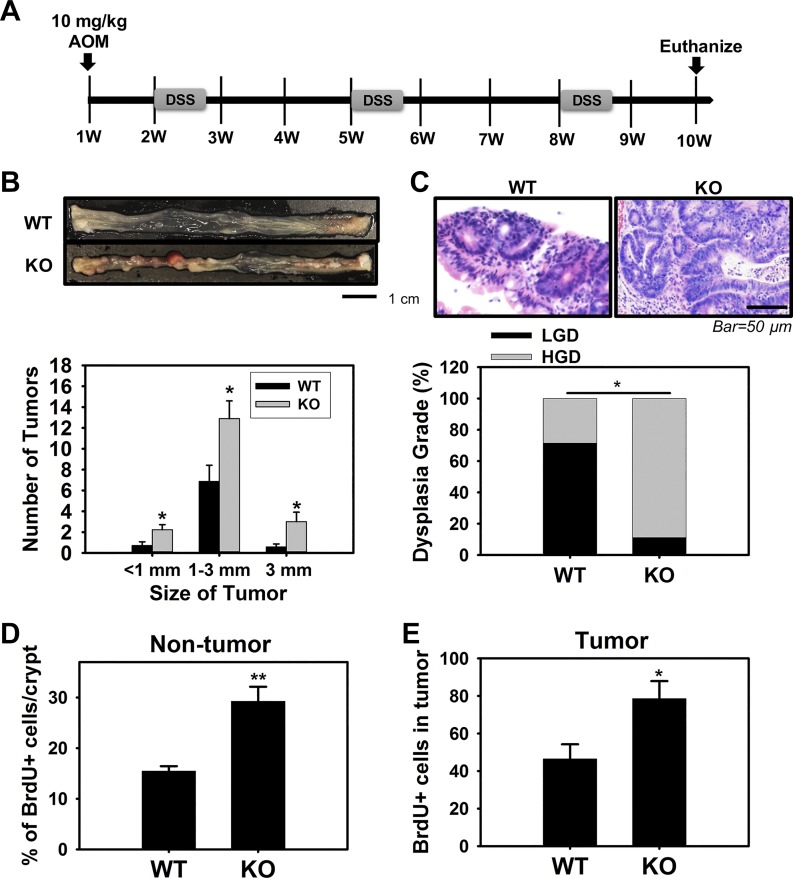
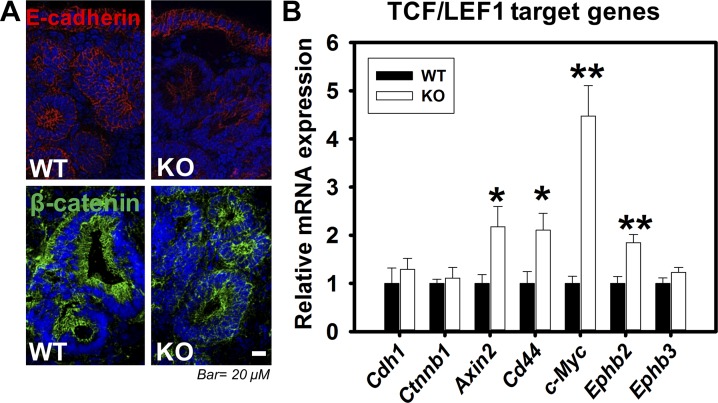
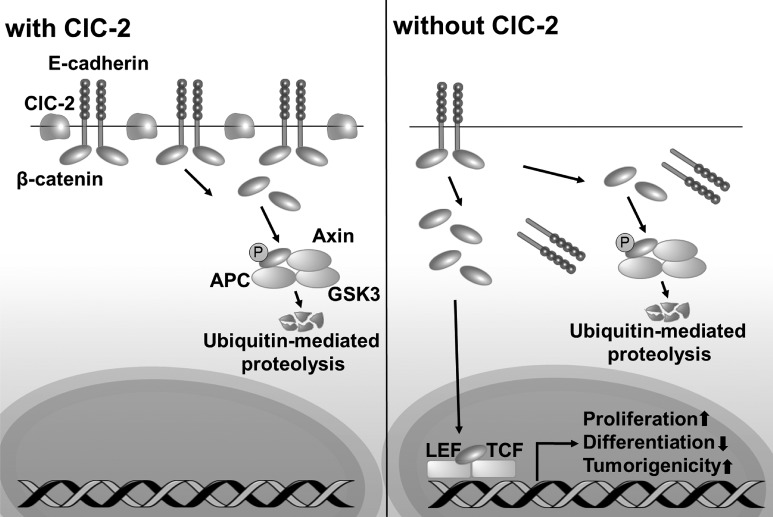
Adherens junctions (AJs), together with tight junctions (TJs), form an apical junctional complex that regulates intestinal epithelial cell-to-cell adherence and barrier homeostasis. Within the AJ, membrane-bound E-cadherin binds β-catenin, which functions as an essential intracellular signaling molecule. We have previously identified a novel protein in the region of the apical junction complex, chloride channel protein-2 (ClC-2), that we have used to study TJ regulation. In this study, we investigated the possible effects of ClC-2 on the regulation of AJs in intestinal mucosal epithelial homeostasis and tumorigenicity. Mucosal homeostasis and junctional proteins were examined in wild-type (WT) and ClC-2 knockout (KO) mice as well as associated colonoids. Tumorigenicity and AJ-associated signaling were evaluated in a murine colitis-associated tumor model and in a colorectal cancer cell line (HT-29). Colonic tissues from ClC-2 KO mice had altered ultrastructural morphology of intercellular junctions with reduced colonocyte differentiation, whereas jejunal tissues had minimal changes. Colonic crypts from ClC-2 KO mice had significantly higher numbers of less-differentiated forms of colonoids compared with WT. Furthermore, the absence of ClC-2 resulted in redistribution of AJ proteins and increased β-catenin activity. Downregulation of ClC-2 in colorectal cells resulted in significant increases in proliferation associated with disruption of AJs. Colitis-associated tumors in ClC-2 KO mice were significantly increased, associated with β-catenin transcription factor activation. The absence of ClC-2 results in less differentiated colonic crypts and increased tumorigenicity associated with colitis via dysregulation of AJ proteins and activation of β-catenin-associated signaling. NEW & NOTEWORTHY Disruption of adherens junctions in the absence of chloride channel protein-2 revealed critical functions of these junctional structures, including maintenance of colonic homeostasis and differentiation as well as driving tumorigenicity by regulating β-catenin signaling.
Keywords: adherens junction; chloride channel ClC-2; colonic homeostasis; colonic tumorigenicity; colonoids.
Figures
Similar articles
-
Expression of tight and adherens junction proteins in ulcerative colitis associated colorectal carcinoma: upregulation of claudin-1, claudin-3, claudin-4, and beta-catenin.Int J Colorectal Dis. 2009 Apr;24(4):361-8. doi: 10.1007/s00384-009-0653-y. Epub 2009 Jan 29. Int J Colorectal Dis. 2009. PMID: 19184060
-
Role of Janus kinase 3 in mucosal differentiation and predisposition to colitis.J Biol Chem. 2013 Nov 1;288(44):31795-806. doi: 10.1074/jbc.M113.504126. Epub 2013 Sep 17. J Biol Chem. 2013. PMID: 24045942 Free PMC article.
-
Overexpression of CD97 in intestinal epithelial cells of transgenic mice attenuates colitis by strengthening adherens junctions.PLoS One. 2010 Jan 13;5(1):e8507. doi: 10.1371/journal.pone.0008507. PLoS One. 2010. PMID: 20084281 Free PMC article.
-
Trans-Compartmental Regulation of Tight Junction Barrier Function.Tissue Barriers. 2023 Oct 2;11(4):2133880. doi: 10.1080/21688370.2022.2133880. Epub 2022 Oct 11. Tissue Barriers. 2023. PMID: 36220768 Free PMC article. Review.
-
N-cadherin-based adherens junction regulates the maintenance, proliferation, and differentiation of neural progenitor cells during development.Cell Adh Migr. 2015;9(3):183-92. doi: 10.1080/19336918.2015.1005466. Epub 2015 Apr 14. Cell Adh Migr. 2015. PMID: 25869655 Free PMC article. Review.
Cited by
-
Loss of ERβ in Aging LXRαβ Knockout Mice Leads to Colitis.Int J Mol Sci. 2023 Aug 5;24(15):12461. doi: 10.3390/ijms241512461. Int J Mol Sci. 2023. PMID: 37569842 Free PMC article.
-
How Dysregulated Ion Channels and Transporters Take a Hand in Esophageal, Liver, and Colorectal Cancer.Rev Physiol Biochem Pharmacol. 2021;181:129-222. doi: 10.1007/112_2020_41. Rev Physiol Biochem Pharmacol. 2021. PMID: 32875386 Review.
-
Ion channels research in hPSC-RPE cells: bridging benchwork to clinical applications.J Transl Med. 2024 Nov 27;22(1):1073. doi: 10.1186/s12967-024-05769-5. J Transl Med. 2024. PMID: 39604931 Free PMC article. Review.
-
Inhibition of ClC-5 suppresses proliferation and induces apoptosis in cholangiocarcinoma cells through the Wnt/β-catenin signaling pathway.BMB Rep. 2022 Jun;55(6):299-304. doi: 10.5483/BMBRep.2022.55.6.044. BMB Rep. 2022. Retraction in: BMB Rep. 2024 Feb;57(2):123. PMID: 35651328 Free PMC article. Retracted.
-
Pathophysiological role of ion channels and transporters in gastrointestinal mucosal diseases.Cell Mol Life Sci. 2021 Dec;78(24):8109-8125. doi: 10.1007/s00018-021-04011-5. Epub 2021 Nov 15. Cell Mol Life Sci. 2021. PMID: 34778915 Free PMC article. Review.
References
-
- Aust DE, Terdiman JP, Willenbucher RF, Chew K, Ferrell L, Florendo C, Molinaro-Clark A, Baretton GB, Lohrs U, Waldman FM. Altered distribution of beta-catenin, and its binding proteins E-cadherin and APC, in ulcerative colitis-related colorectal cancers. Mod Pathol 14: 29–39, 2001. doi:10.1038/modpathol.3880253. - DOI - PubMed
-
- Barrett CW, Singh K, Motley AK, Lintel MK, Matafonova E, Bradley AM, Ning W, Poindexter SV, Parang B, Reddy VK, Chaturvedi R, Fingleton BM, Washington MK, Wilson KT, Davies SS, Hill KE, Burk RF, Williams CS. Dietary selenium deficiency exacerbates DSS-induced epithelial injury and AOM/DSS-induced tumorigenesis. PLoS One 8: e67845, 2013. doi:10.1371/journal.pone.0067845. - DOI - PMC - PubMed
-
- Boivin GP, Washington K, Yang K, Ward JM, Pretlow TP, Russell R, Besselsen DG, Godfrey VL, Doetschman T, Dove WF, Pitot HC, Halberg RB, Itzkowitz SH, Groden J, Coffey RJ. Pathology of mouse models of intestinal cancer: consensus report and recommendations. Gastroenterology 124: 762–777, 2003. doi:10.1053/gast.2003.50094. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials