

Bead-linked transposomes enable a normalization-free workflow for NGS library preparation
- PMID: 30285621
- PMCID: PMC6167868
- DOI: 10.1186/s12864-018-5096-9
Bead-linked transposomes enable a normalization-free workflow for NGS library preparation
Abstract
Background: Transposome-based technologies have enabled the streamlined production of sequencer-ready DNA libraries; however, current methods are highly sensitive to the amount and quality of input nucleic acid.
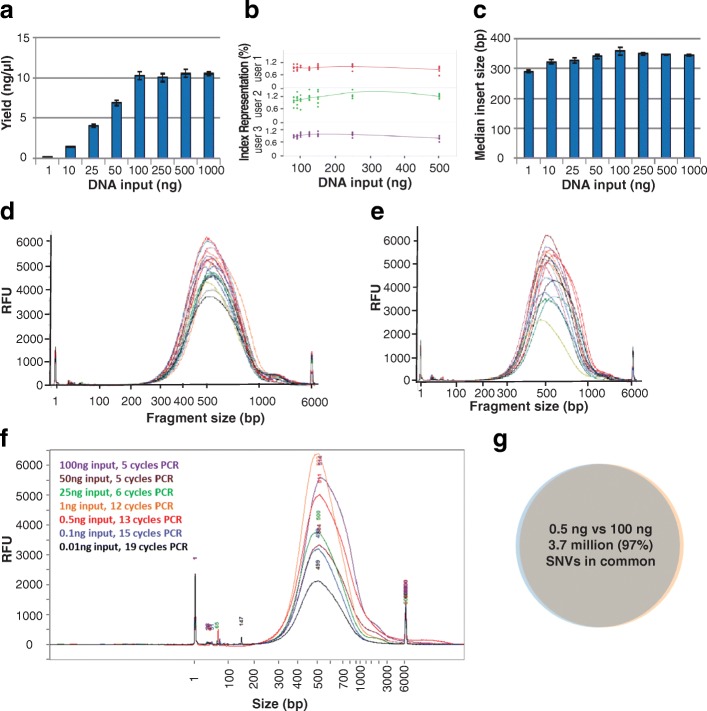
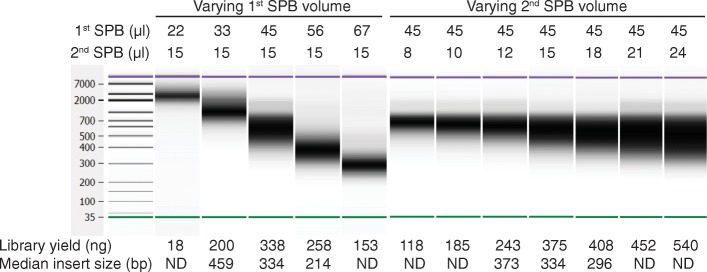
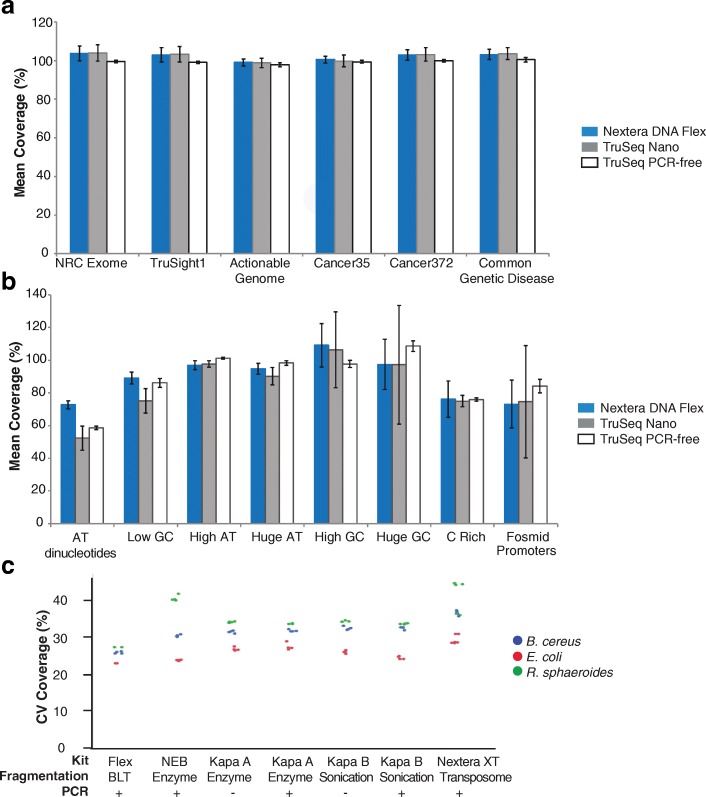
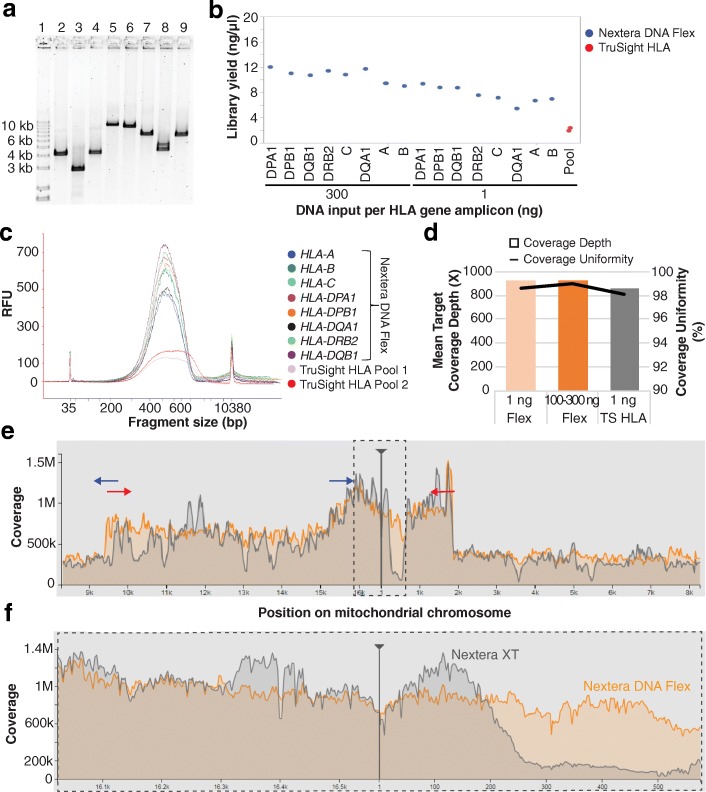
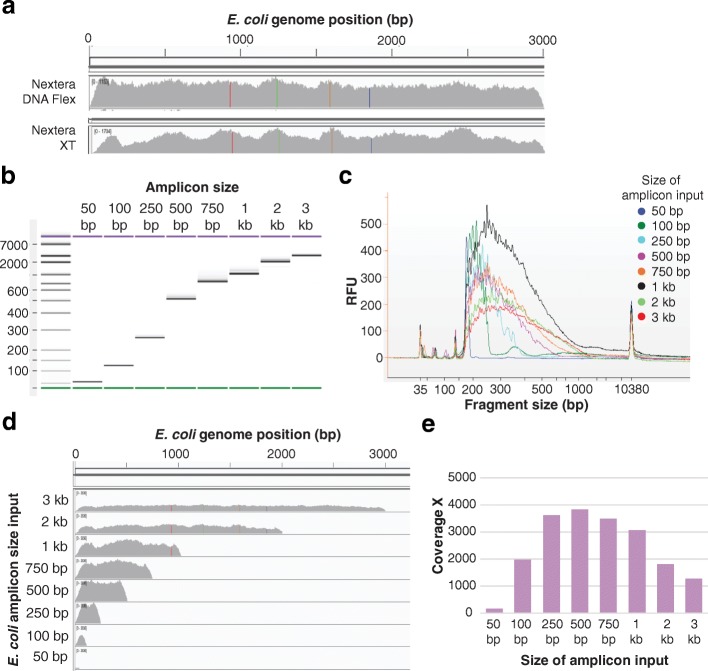
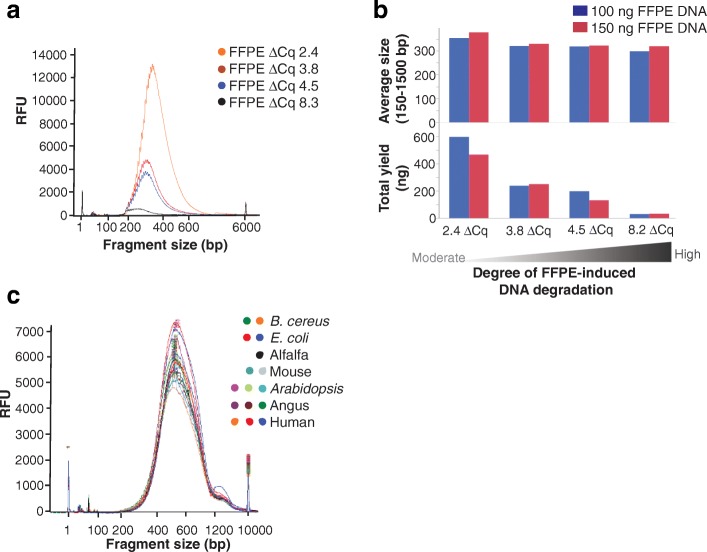
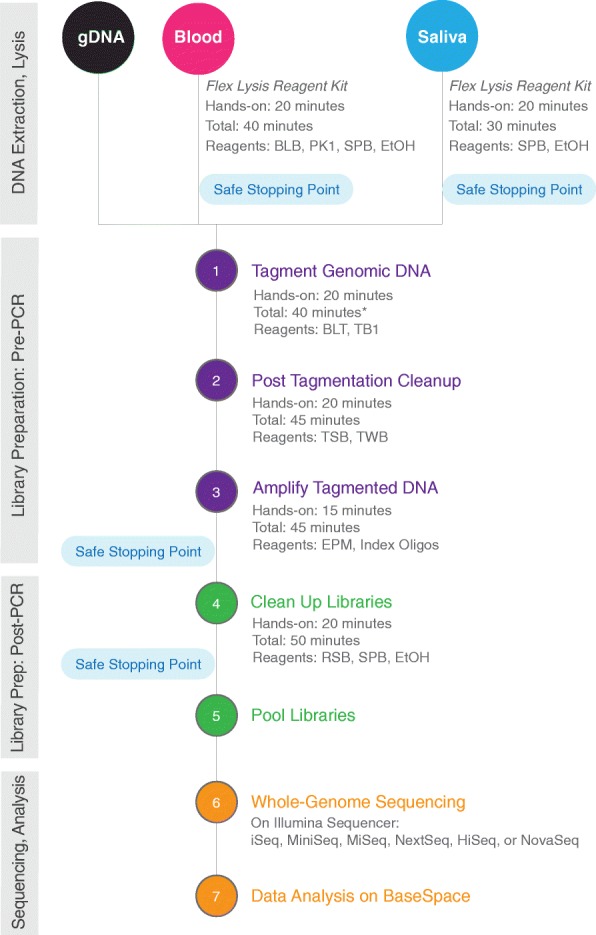
Results: We describe a new library preparation technology (Nextera DNA Flex) that utilizes a known concentration of transposomes conjugated directly to beads to bind a fixed amount of DNA, and enables direct input of blood and saliva using an integrated extraction protocol. We further report results from libraries generated outside the standard parameters of the workflow, highlighting novel applications for Nextera DNA Flex, including human genome builds and variant calling from below 1 ng DNA input, customization of insert size, and preparation of libraries from short fragments and severely degraded FFPE samples. Using this bead-linked library preparation method, library yield saturation was observed at an input amount of 100 ng. Preparation of libraries from a range of species with varying GC levels demonstrated uniform coverage of small genomes. For large and complex genomes, coverage across the genome, including difficult regions, was improved compared with other library preparation methods. Libraries were successfully generated from amplicons of varying sizes (from 50 bp to 11 kb), however, a decrease in efficiency was observed for amplicons smaller than 250 bp. This library preparation method was also compatible with poor-quality DNA samples, with sequenceable libraries prepared from formalin-fixed paraffin-embedded samples with varying levels of degradation.
Conclusions: In contrast to solution-based library preparation, this bead-based technology produces a normalized, sequencing-ready library for a wide range of DNA input types and amounts, largely obviating the need for DNA quantitation. The robustness of this bead-based library preparation kit and flexibility of input DNA facilitates application across a wide range of fields.
Keywords: Library preparation; Next-generation sequencing; Transposome.
Conflict of interest statement
Ethics approval and consent to participate
Human blood and saliva samples were collected from healthy volunteers of at least 18 years of age that provided written consent to use of their samples for research. The study was approved by an independent Institutional Review Board (IRB), Quorum Review IRB (
Consent for publication
Not applicable.
Competing interests
All authors are employees of and hold stock in Illumina, Inc.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- Kim S, Scheffler K, Halpern AL, Bekritsky MA, Noh E, Källberg M, Chen X, Beyter D, Krusche P, Saunders CT. bioRxiv. 2017. Strelka2: fast and accurate variant calling for clinical sequencing applications. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
