

Urea cycle disorders in India: clinical course, biochemical and genetic investigations, and prenatal testing
- PMID: 30285816
- PMCID: PMC6167905
- DOI: 10.1186/s13023-018-0908-1
Urea cycle disorders in India: clinical course, biochemical and genetic investigations, and prenatal testing
Abstract
Background: Urea cycle disorders (UCDs) are inherited metabolic disorders that present with hyperammonemia, and cause significant mortality and morbidity in infants and children. These disorders are not well reported in the Indian population, due to lack of a thorough study of the clinical and molecular profile.
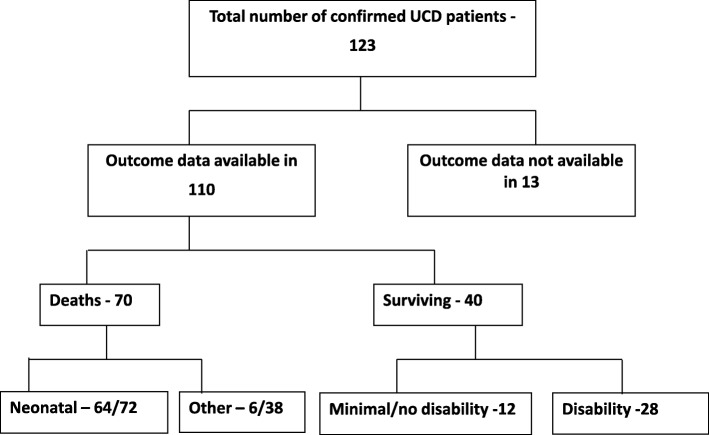
Results: We present data from two major metabolic centres in India, including 123 cases of various UCDs. The majority of them (72/123, 58%) presented in the neonatal period (before 30 days of age) with 88% on or before day 7 of life (classical presentation), and had a high mortality (64/72, 88%). Citrullinemia type 1 was the most common UCD, observed in 61/123 patients. Ornithine transcarbamylase (OTC) deficiency was the next most common, seen in 24 cases. Argininosuccinic aciduria was diagnosed in 20 cases. Deficiencies of arginase, N-acetylglutamate synthase, carbamoyl phosphate synthetase, citrin, and lysinuric protein intolerance were also observed. Molecular genetic analysis revealed two common ASS1 mutations: c.470G > A (p.Arg157His) and c.1168G > A (p.Gly390Arg) (36 of 55 tested patients). In addition, few recurrent point mutations in ASL gene, and a deletion of the whole OTC gene were also noted. A total of 24 novel mutations were observed in the various genes studied. We observed a poor clinical outcome with an overall all time mortality of 63% (70/110 cases with a known follow-up), and disability in 70% (28/40) among the survivors. Prenatal diagnosis was performed in 30 pregnancies in 25 families, including one pre-implantation genetic diagnosis.
Conclusions: We report the occurrence of UCDs in India and the spectrum that may be different from the rest of the world. Citrullinemia type 1 was the most common UCD observed in the cohort. Increasing awareness amongst clinicians will improve outcomes through early diagnosis and timely treatment. Genetic diagnosis in the proband will enable prenatal/pre-implantation diagnosis in subsequent pregnancies.
Keywords: Argininosuccinic aciduria; Citrullinemia; Hyperammonemia; Mutation; OTC deficiency; Prenatal diagnosis; UCD; Urea cycle.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Consent has been taken from all the authors. No individual patient data including videos, voice recordings or images have been submitted for publication.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- Häberle J, Boddaert N, Burlina A, Chakrapani A, Dixon M, Huemer M, Karall D, Martinelli D, Sanjurjo Crespo P, Santer R, Servais A, Valayannopoulos V, Lindner M, Rubio V, Dionisi-Vici C. Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J Rare Dis. 2012;7:32. doi: 10.1186/1750-1172-7-32. - DOI - PMC - PubMed
-
- Brusilow SW, Maestri NE. Urea cycle disorders: diagnosis, pathophysiology, and therapy. Adv Pediatr Infect Dis. 1996;43:127–170. - PubMed
-
- Wilcken B, Haas M, Joy P, Wiley V, Bowling F, Carpenter K, Christodoulou J, Cowley D, Ellaway C, Fletcher J, Kirk EP, Lewis B, McGill J, Peters H, Pitt J, Ranieri E, Yaplito-Lee J, Boneh A. Expanded newborn screening: outcome in screened and unscreened patients at age 6 years. Pediatrics. 2009;124:e241–e248. doi: 10.1542/peds.2008-0586. - DOI - PubMed
-
- Nettesheim S, Kölker S, Karall D, Häberle J, Posset R, Hoffmann GF, HeinrichB GF, Garbade SF, Arbeitsgemeinschaft für Pädiatrische Stoffwechselstörungen (APS); European registry and network for Intoxication type Metabolic Diseases (E-IMD); Erhebungseinheit für Seltene PädiatrischeErkrankungen in Deutschland (ESPED); Austrian Metabolic Group; Swiss Paediatric Surveillance Unit (SPSU) Incidence, disease onset and short-term outcome in urea cycle disorders -cross-border surveillance in Germany, Austria and Switzerland. Orphanet J Rare Dis. 2017;12(1):111. doi: 10.1186/s13023-017-0661-x. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
