

Prediction of the Closed Conformation and Insights into the Mechanism of the Membrane Enzyme LpxR
- PMID: 30287112
- PMCID: PMC6260217
- DOI: 10.1016/j.bpj.2018.09.002
Prediction of the Closed Conformation and Insights into the Mechanism of the Membrane Enzyme LpxR
Abstract
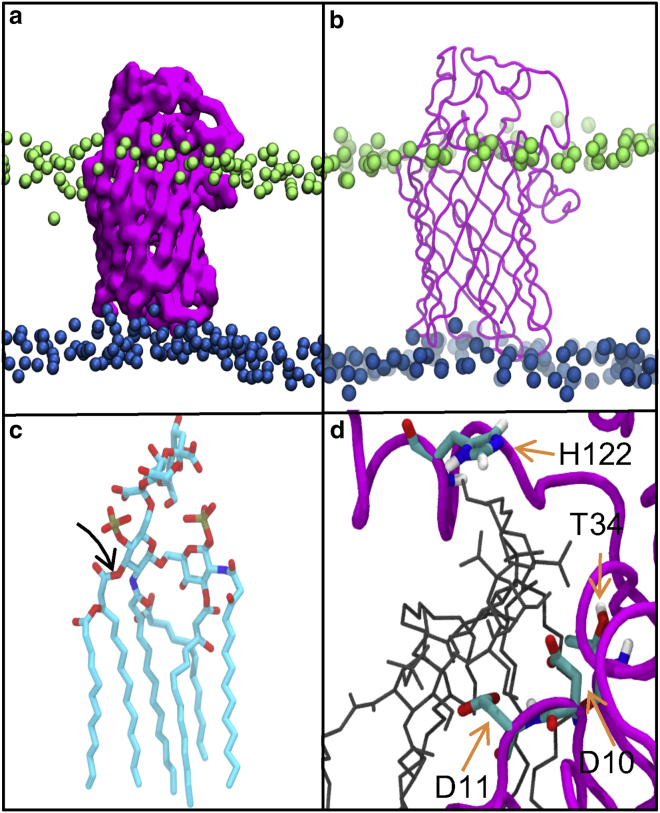
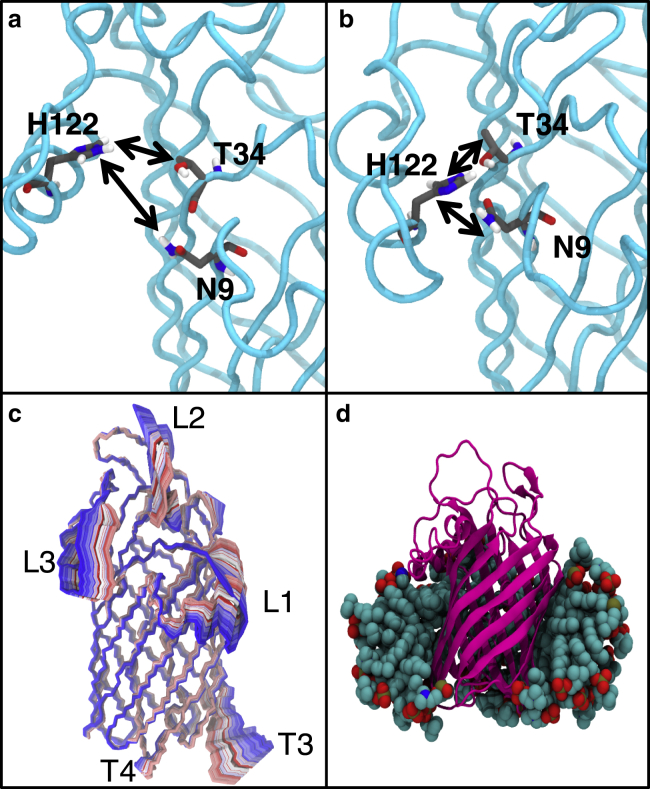
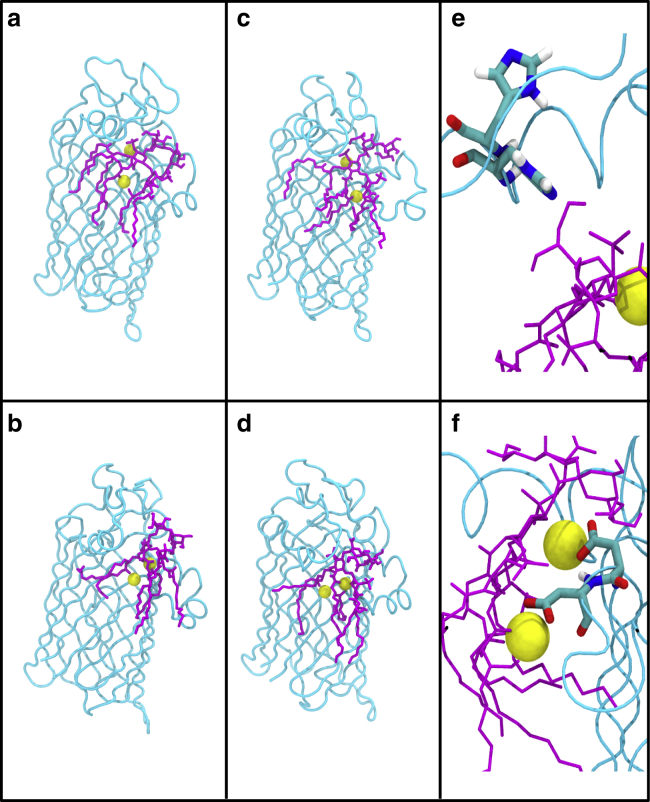
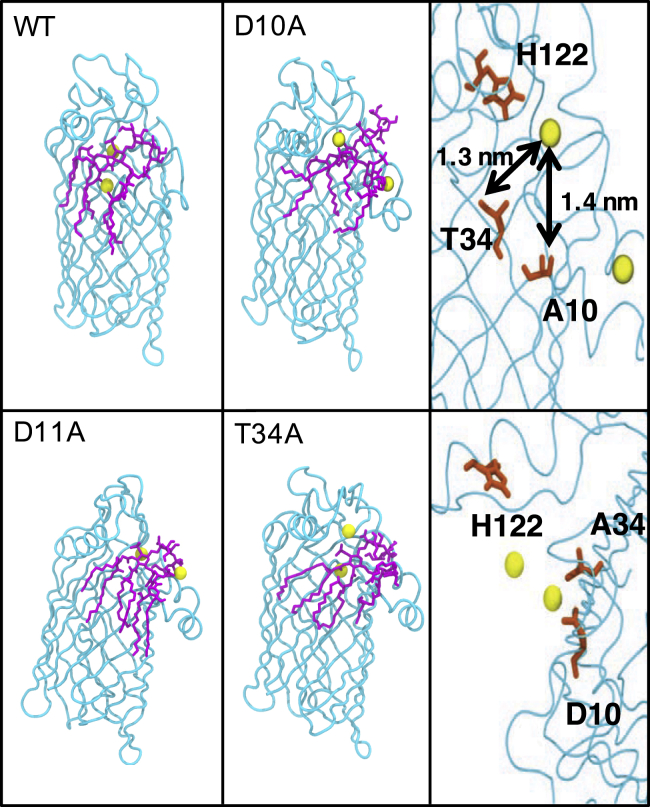
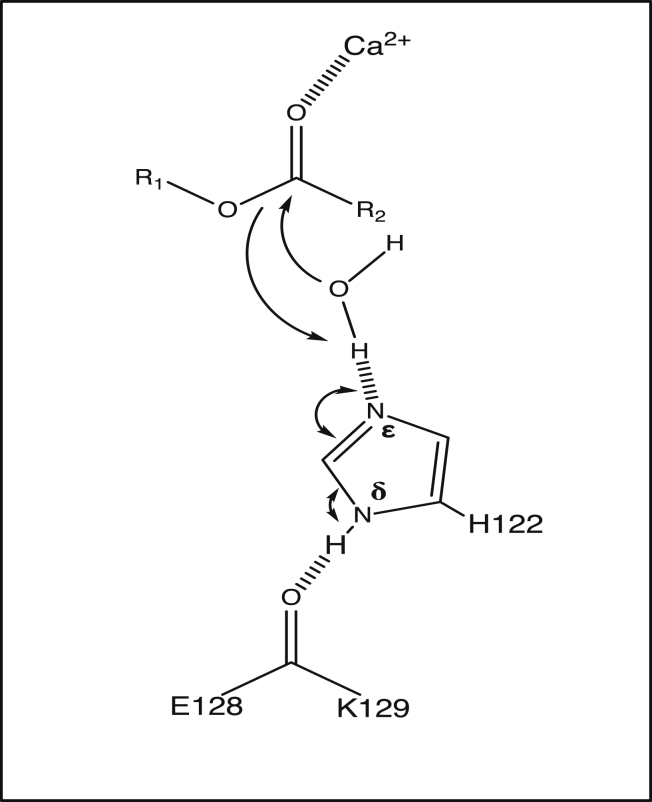
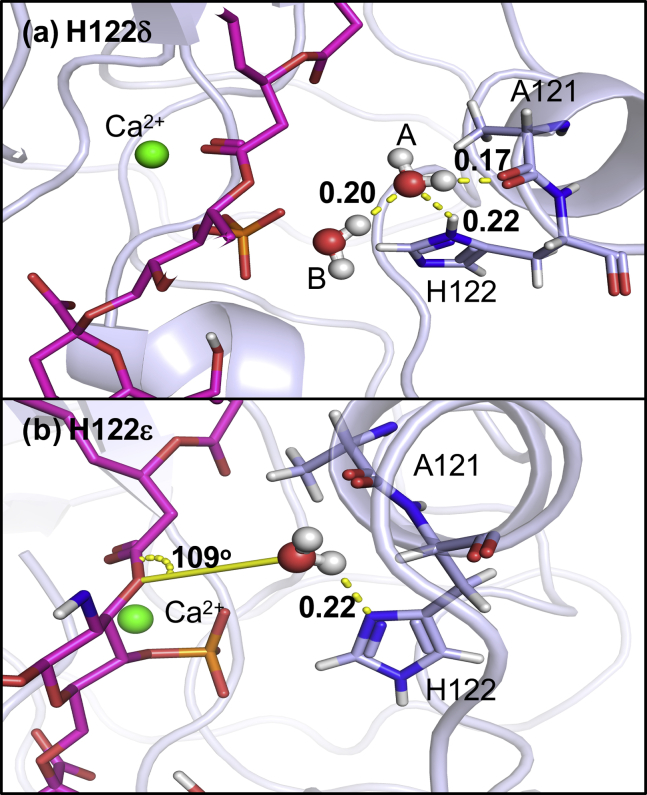
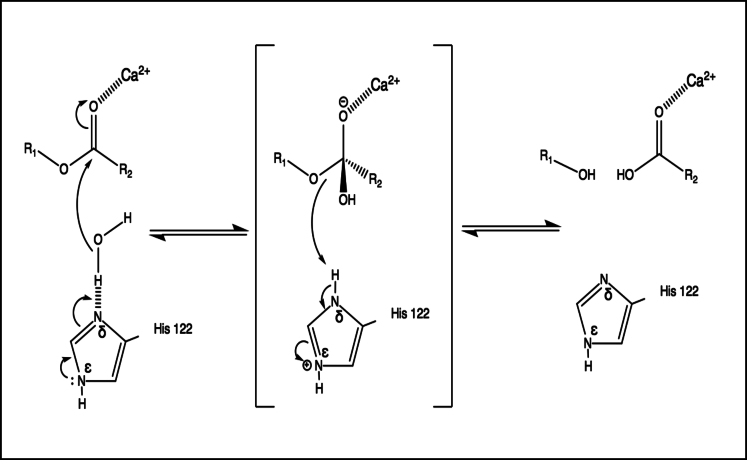
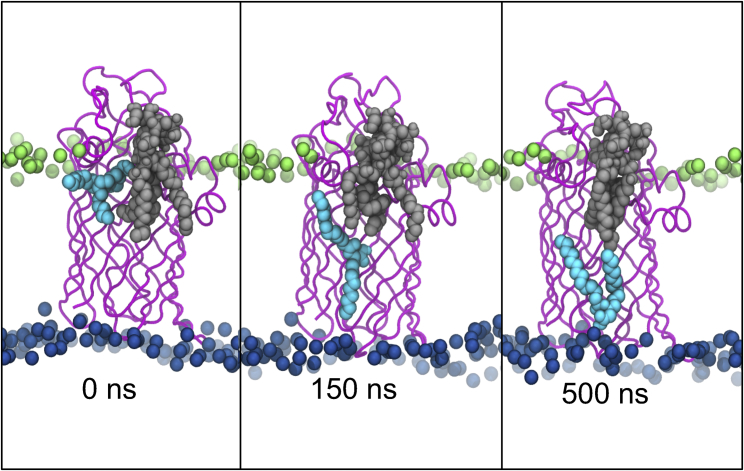
Covalent modification of outer membrane lipids of Gram-negative bacteria can impact the ability of the bacterium to develop resistance to antibiotics as well as modulating the immune response of the host. The enzyme LpxR from Salmonella typhimurium is known to deacylate lipopolysaccharide molecules of the outer membrane; however, the mechanism of action is unknown. Here, we employ molecular dynamics and Monte Carlo simulations to study the conformational dynamics and substrate binding of LpxR in representative outer membrane models as well as detergent micelles. We examine the roles of conserved residues and provide an understanding of how LpxR binds its substrate. Our simulations predict that the catalytic H122 must be Nε-protonated for a single water molecule to occupy the space between it and the scissile bond, with a free binding energy of -8.5 kcal mol-1. Furthermore, simulations of the protein within a micelle enable us to predict the structure of the putative "closed" protein. Our results highlight the need for including dynamics, a representative environment, and the consideration of multiple tautomeric and rotameric states of key residues in mechanistic studies; static structures alone do not tell the full story.
Copyright © 2018 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Hwang P.M., Kay L.E. Solution structure and dynamics of integral membrane proteins by NMR: a case study involving the enzyme PagP. Methods Enzymol. 2005;394:335–350. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
