

Simulations of the regulatory ACT domain of human phenylalanine hydroxylase (PAH) unveil its mechanism of phenylalanine binding
- PMID: 30287685
- PMCID: PMC6314134
- DOI: 10.1074/jbc.RA118.004909
Simulations of the regulatory ACT domain of human phenylalanine hydroxylase (PAH) unveil its mechanism of phenylalanine binding
Abstract
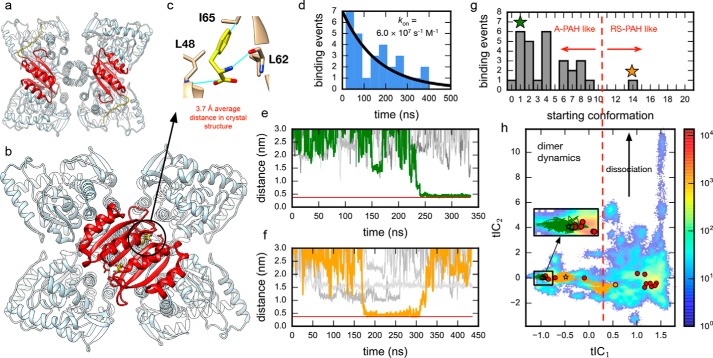
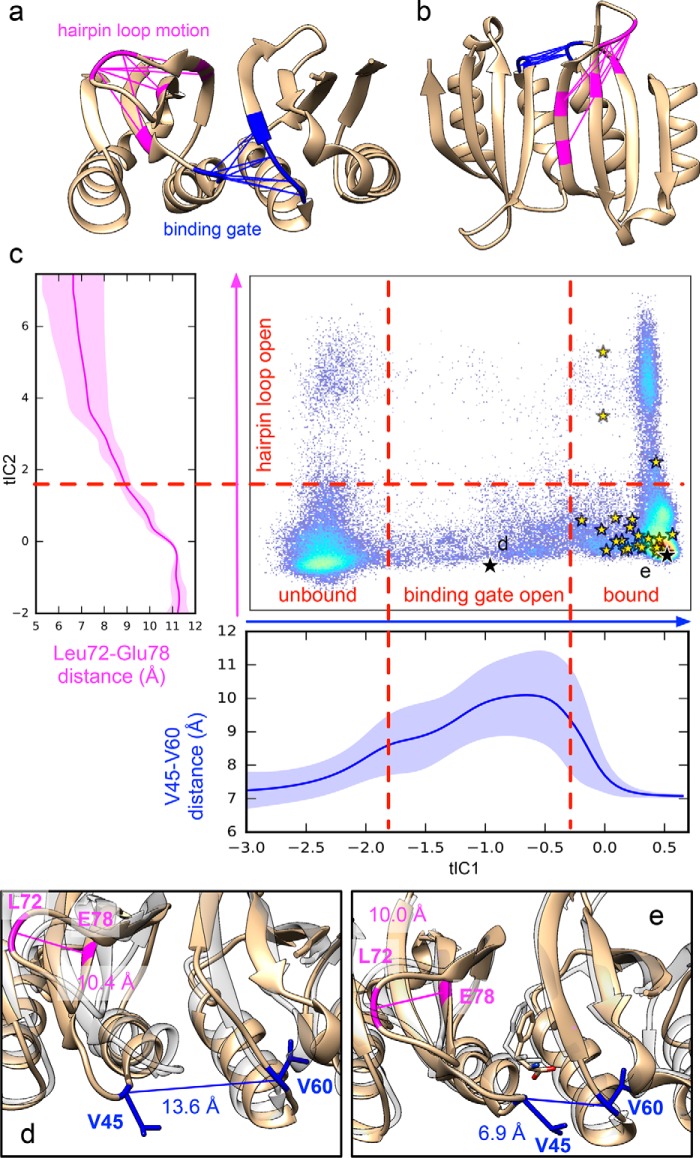
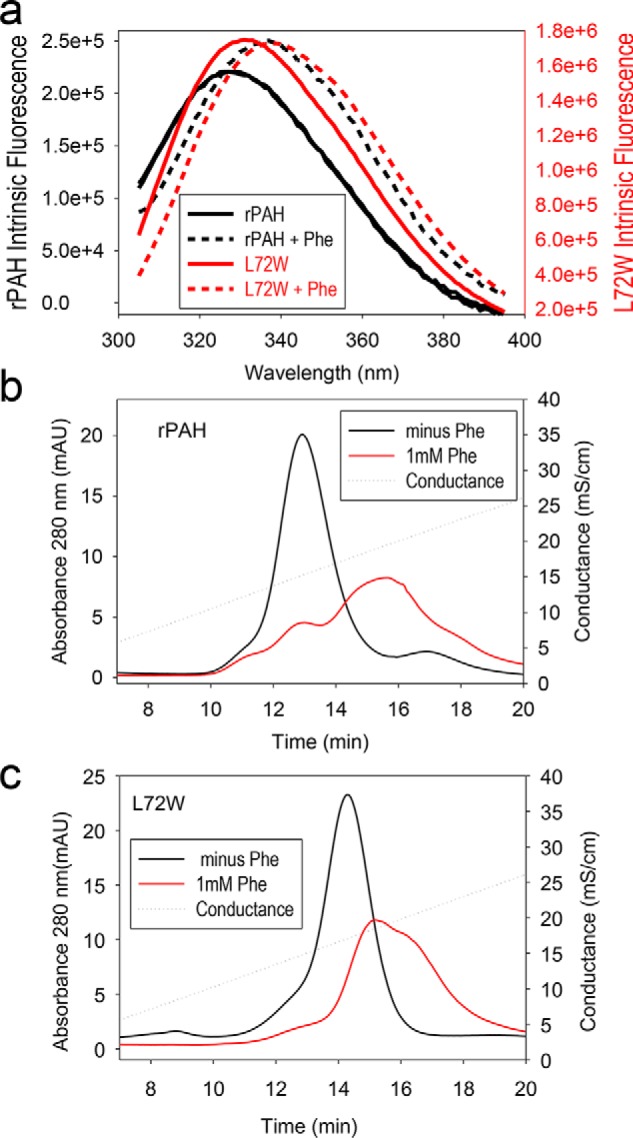
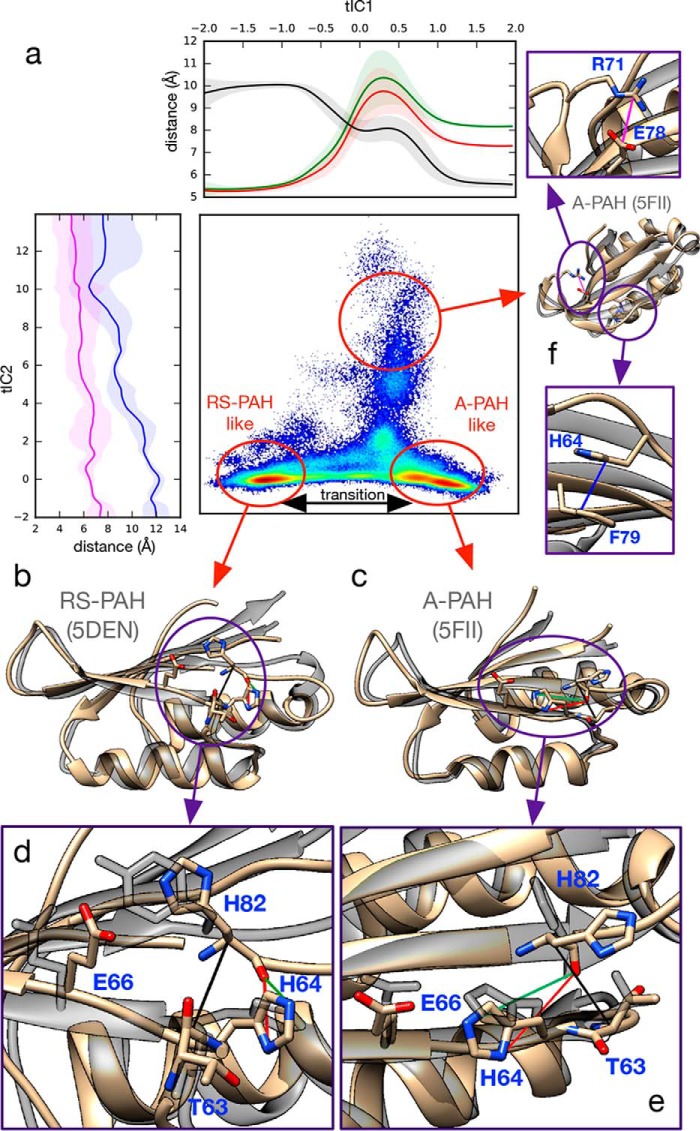
Phenylalanine hydroxylase (PAH) regulates phenylalanine (Phe) levels in mammals to prevent neurotoxicity resulting from high Phe concentrations as observed in genetic disorders leading to hyperphenylalaninemia and phenylketonuria. PAH senses elevated Phe concentrations by transient allosteric Phe binding to a protein-protein interface between ACT domains of different subunits in a PAH tetramer. This interface is present in an activated PAH (A-PAH) tetramer and absent in a resting-state PAH (RS-PAH) tetramer. To investigate this allosteric sensing mechanism, here we used the GROMACS molecular dynamics simulation suite on the Folding@home computing platform to perform extensive molecular simulations and Markov state model (MSM) analysis of Phe binding to ACT domain dimers. These simulations strongly implicated a conformational selection mechanism for Phe association with ACT domain dimers and revealed protein motions that act as a gating mechanism for Phe binding. The MSMs also illuminate a highly mobile hairpin loop, consistent with experimental findings also presented here that the PAH variant L72W does not shift the PAH structural equilibrium toward the activated state. Finally, simulations of ACT domain monomers are presented, in which spontaneous transitions between resting-state and activated conformations are observed, also consistent with a mechanism of conformational selection. These mechanistic details provide detailed insight into the regulation of PAH activation and provide testable hypotheses for the development of new allosteric effectors to correct structural and functional defects in PAH.
Keywords: Markov state models; allosteric regulation; binding pathways; conformational change; conformational selection; kinetics; ligand binding kinetics; ligand-binding protein; molecular dynamics; molecular simulation; phenylalanine hydroxylase; phenylketonuria.
© 2018 Ge et al.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures
References
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
