

TGF-β1 programmed myeloid-derived suppressor cells (MDSC) acquire immune-stimulating and tumor killing activity capable of rejecting established tumors in combination with radiotherapy
- PMID: 30288358
- PMCID: PMC6169570
- DOI: 10.1080/2162402X.2018.1490853
TGF-β1 programmed myeloid-derived suppressor cells (MDSC) acquire immune-stimulating and tumor killing activity capable of rejecting established tumors in combination with radiotherapy
Abstract
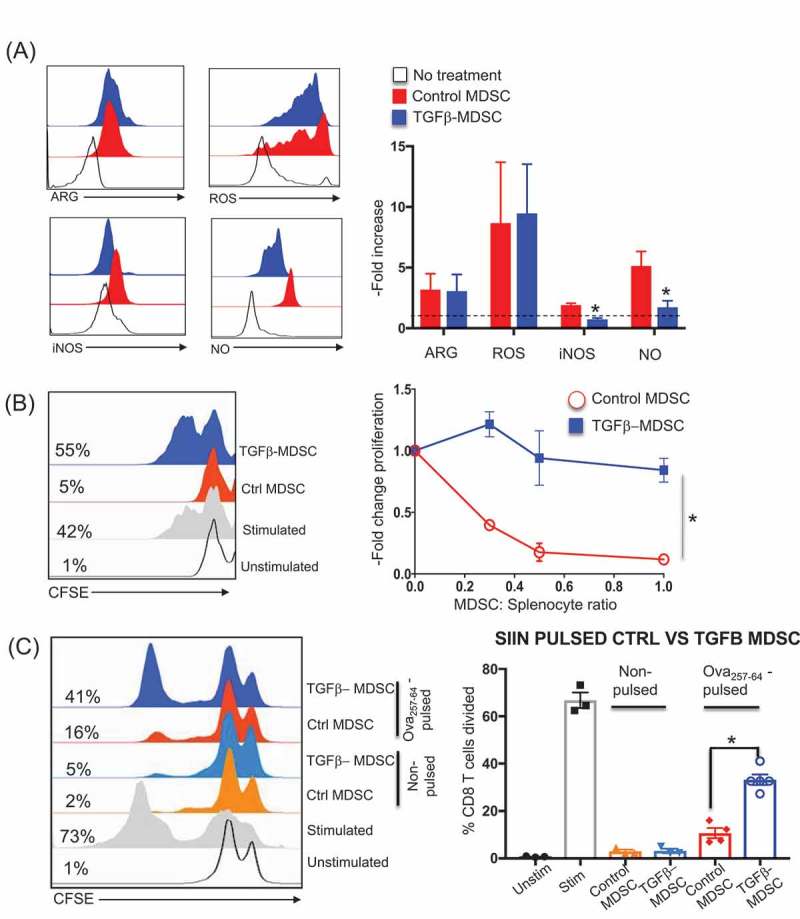
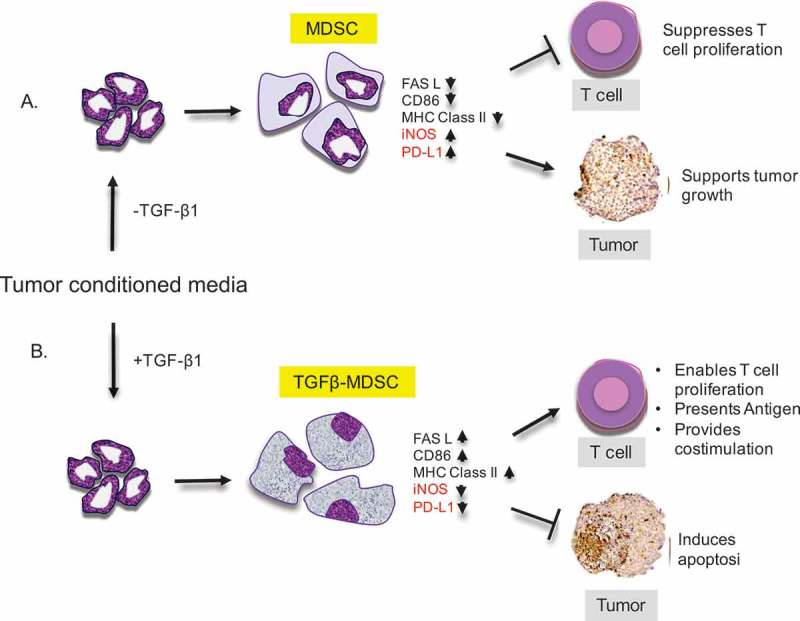
Cancer-induced myeloid-derived suppressor cells (MDSC) play an important role in tumor immune evasion. MDSC programming or polarization has been proposed as a strategy for leveraging the developmental plasticity of myeloid cells to reverse MDSC immune suppressive functions, or cause them to acquire anti-tumor activity. While MDSC derived ex vivo from murine bone marrow precursor cells with tumor-conditioned medium efficiently suppressed T cell proliferation, MDSC derived from conditioned medium in presence of TGF-β1 (TGFβ-MDSC) acquired a novel immune-stimulatory phenotype, losing the ability to inhibit T cell proliferation and acquiring enhanced antigen-presenting capability. Altered immune function was associated with SMAD-2 dependent upregulation of maturation and costimulatory molecules, and downregulation of inducible nitric oxide synthase (iNOS), an effector mechanism of immunosuppression. TGFβ-MDSC also upregulated FAS-ligand expression, leading to FAS-dependent killing of murine human papillomavirus (HPV)-associated head and neck cancer cells and tumor spheroids in vitro and anti-tumor activity in vivo. Radiation upregulated FAS expression on tumor cells, and the combination of radiotherapy and intratumoral injection of TGFβ-MDSC strongly enhanced class I expression on tumor cells and induction of HPV E7 tetramer-positive CD8 + T cells, leading to clearance of established tumors and long-term survival. TGFβ-MDSC derived from human PBMC with tumor conditioned medium also lost immunosuppressive function and acquired tumor-killing activity. Thus, TGFβ1 mediated programming of nascent MDSC leads to a potent anti-tumor phenotype potentially suitable for adoptive immunotherapy.
Keywords: CD86; MDSC; SMAD2; TGF-beta; adoptive cellular therapy; caspase-3; myeloid-derived suppressor cells (written out); radiotherapy; tumor killing.
Figures





References
-
- Rosenberg SA. Decade in review—cancer immunotherapy: entering the mainstream of cancer treatment. Nat Rev Clin Oncol. Internet. 2014; 11: 630–632. Available from: http://www.nature.com/doifinder/10.1038/nrclinonc.2014.174 doi: 10.1038/nrclinonc.2014.174. - DOI - DOI - PMC - PubMed
-
- Topalian SL, Weiner GJ, Pardoll DM. Cancer immunotherapy comes of age. J Clin Oncol. Internet. 2011; 29: 4828–4836. Available from: http://jco.ascopubs.org/content/29/36/4828.short doi: 10.1200/JCO.2011.38.0899. - DOI - PMC - PubMed
-
- Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer. Internet. 2012; 12: 237–251. Available from: http://www.nature.com/doifinder/10.1038/nrc3237 doi: 10.1038/nrc3237. - DOI - DOI - PMC - PubMed
-
- Postow MA, Callahan MK, Wolchok JD. Immune Checkpoint Blockade in Cancer Therapy. J Clin Oncol. Internet. 2015; 33: JCO.2014.59.4358- Available from: http://jco.ascopubs.org/content/early/2015/01/20/JCO.2014.59.4358.full doi: 10.1200/JCO.2014.59.4358. - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous