

Rapid therapeutic advances in CFTR modulator science
- PMID: 30289627
- PMCID: PMC6585954
- DOI: 10.1002/ppul.24157
Rapid therapeutic advances in CFTR modulator science
Abstract
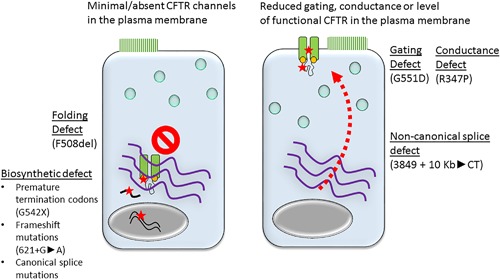
Cystic fibrosis (CF) is an autosomal recessive genetic disease caused by variants in the gene encoding the cystic fibrosis transmembrane conduction regulator (CFTR) protein. Loss of CFTR function disrupts chloride, bicarbonate and regulation of sodium transport, producing a cascade of mucus obstruction, inflammation, pulmonary infection, and ultimately damage in numerous organs. Established CF therapies treat the downstream consequences of CFTR dysfunction and have led to steady improvements in patient survival. A class of drugs termed CFTR modulators has recently entered the CF therapeutic landscape. These drugs differ fundamentally from prior therapies in that they aim to improve the function of disease-causing CFTR variants. This review summarizes the science behind CFTR modulators, including their targets, mechanism of action, clinical benefit, and future directions in the field. CFTR modulators have dramatically changed how CF is treated, validated CFTR as a therapeutic target, and opened the door to truly personalized therapies and treatment regimens.
Keywords: CFTR; cystic fibrosis; ion transport; modulator.
© 2018 Wiley Periodicals, Inc.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
-
- Spielberg DR, Clancy JP. Cystic fibrosis and its management through established and emerging therapies. Annu Rev Genomics Hum Genet. 2016;17:155–175. - PubMed
-
- Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med. 2005;352:1992–2001. - PubMed
-
- Riordan JR, Rommens JM, Kerem B, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. - PubMed
-
- Fuchs HJ, Borowitz DS, Christiansen DH, et al. Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. The Pulmozyme Study Group. N Engl J Med. 1994;331:637–642. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
