

Phenotype-Specific Enrichment of Mendelian Disorder Genes near GWAS Regions across 62 Complex Traits
- PMID: 30290150
- PMCID: PMC6174356
- DOI: 10.1016/j.ajhg.2018.08.017
Phenotype-Specific Enrichment of Mendelian Disorder Genes near GWAS Regions across 62 Complex Traits
Abstract
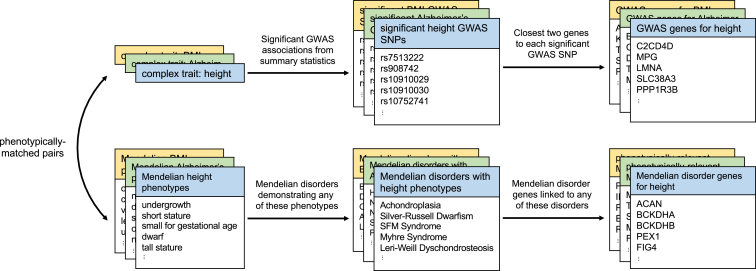
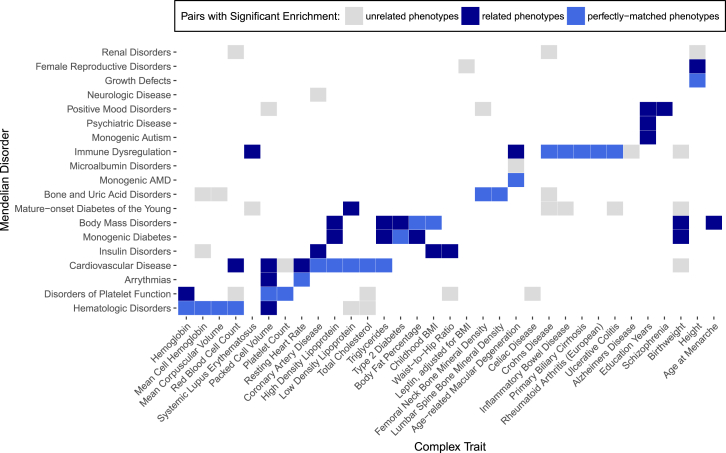
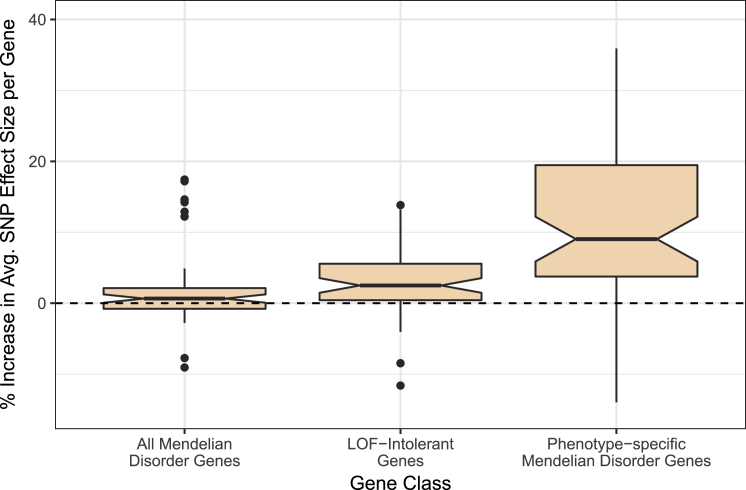
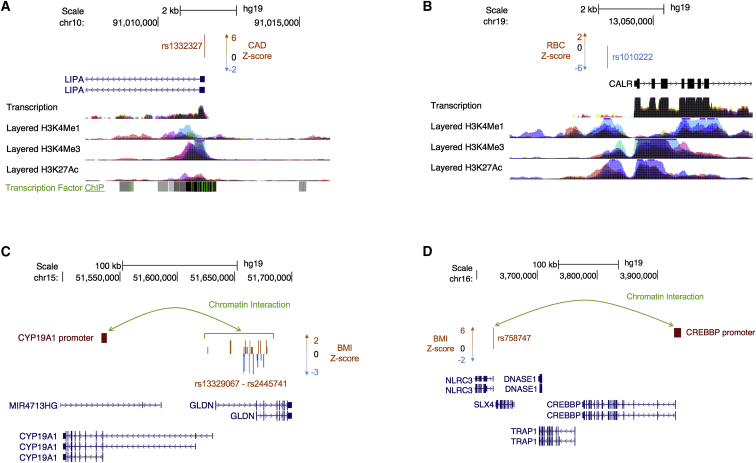
Although recent studies provide evidence for a common genetic basis between complex traits and Mendelian disorders, a thorough quantification of their overlap in a phenotype-specific manner remains elusive. Here, we have quantified the overlap of genes identified through large-scale genome-wide association studies (GWASs) for 62 complex traits and diseases with genes containing mutations known to cause 20 broad categories of Mendelian disorders. We identified a significant enrichment of genes linked to phenotypically matched Mendelian disorders in GWAS gene sets; of the total 1,240 comparisons, a higher proportion of phenotypically matched or related pairs (n = 50 of 92 [54%]) than phenotypically unmatched pairs (n = 27 of 1,148 [2%]) demonstrated significant overlap, confirming a phenotype-specific enrichment pattern. Further, we observed elevated GWAS effect sizes near genes linked to phenotypically matched Mendelian disorders. Finally, we report examples of GWAS variants localized at the transcription start site or physically interacting with the promoters of genes linked to phenotypically matched Mendelian disorders. Our results are consistent with the hypothesis that genes that are disrupted in Mendelian disorders are dysregulated by non-coding variants in complex traits and demonstrate how leveraging findings from related Mendelian disorders and functional genomic datasets can prioritize genes that are putatively dysregulated by local and distal non-coding GWAS variants.
Keywords: GWAS; Hi-C; Mendelian; body mass index; common disease; complex traits; monogenic; polygenic; statistical genetics.
Copyright © 2018 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Cutting G.R., Kasch L.M., Rosenstein B.J., Zielenski J., Tsui L.C., Antonarakis S.E., Kazazian H.H., Jr. A cluster of cystic fibrosis mutations in the first nucleotide-binding fold of the cystic fibrosis conductance regulator protein. Nature. 1990;346:366–369. - PubMed
Publication types
MeSH terms
Grants and funding
- R01 DK093757/DK/NIDDK NIH HHS/United States
- T32 MH073526/MH/NIMH NIH HHS/United States
- U01 CA194393/CA/NCI NIH HHS/United States
- R01 HG006399/HG/NHGRI NIH HHS/United States
- T32 HG002536/HG/NHGRI NIH HHS/United States
- P01 HL028481/HL/NHLBI NIH HHS/United States
- R01 MH115676/MH/NIMH NIH HHS/United States
- F31 HL142180/HL/NHLBI NIH HHS/United States
- R01 HG009120/HG/NHGRI NIH HHS/United States
- DP5 OD024579/OD/NIH HHS/United States
- T32 LM012424/LM/NLM NIH HHS/United States
- R01 HL095056/HL/NHLBI NIH HHS/United States
- F31 DK118865/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
