

PRKN-regulated mitophagy and cellular senescence during COPD pathogenesis
- PMID: 30290714
- PMCID: PMC6351145
- DOI: 10.1080/15548627.2018.1532259
PRKN-regulated mitophagy and cellular senescence during COPD pathogenesis
Abstract
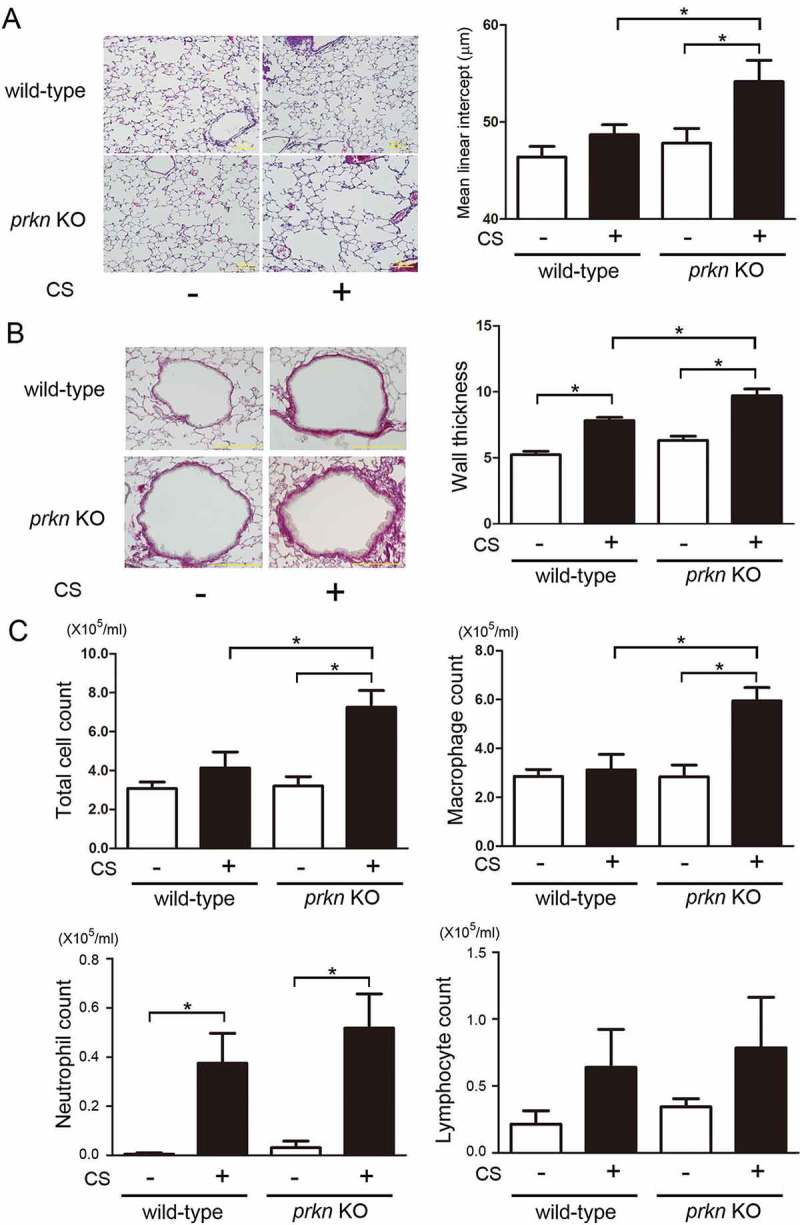
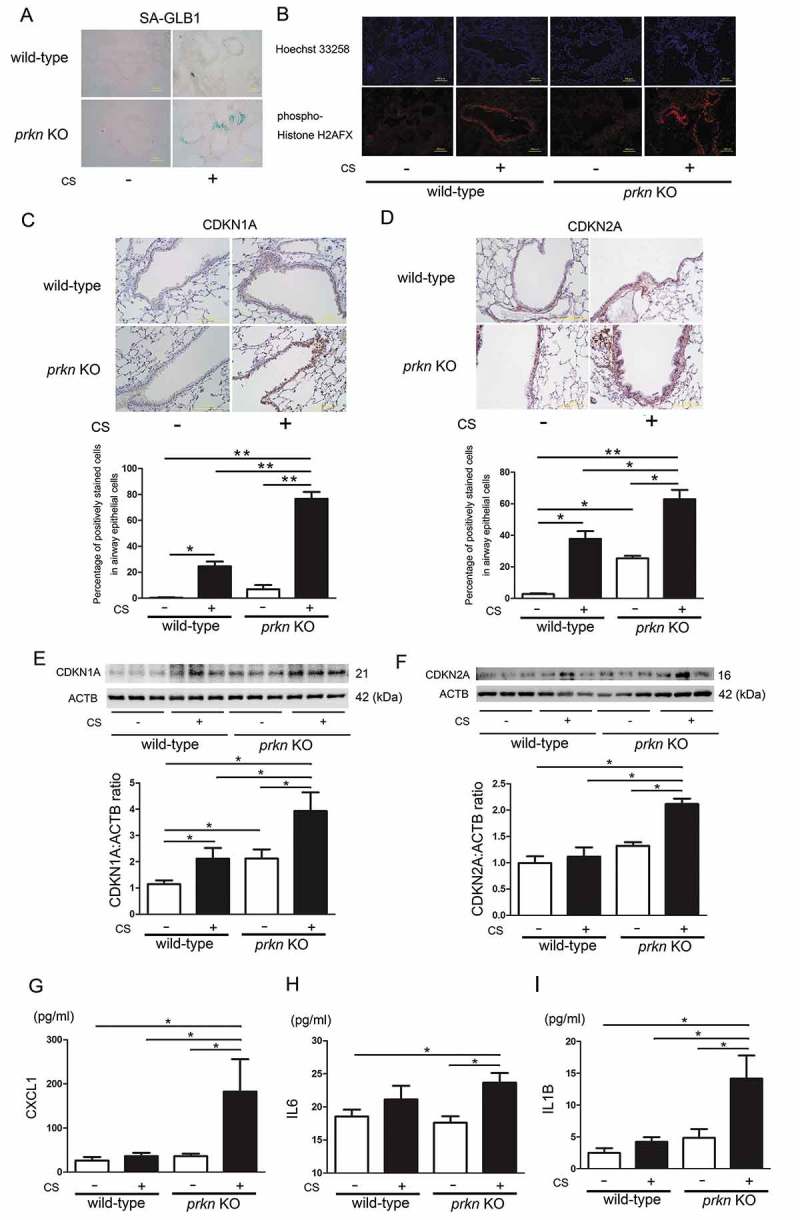
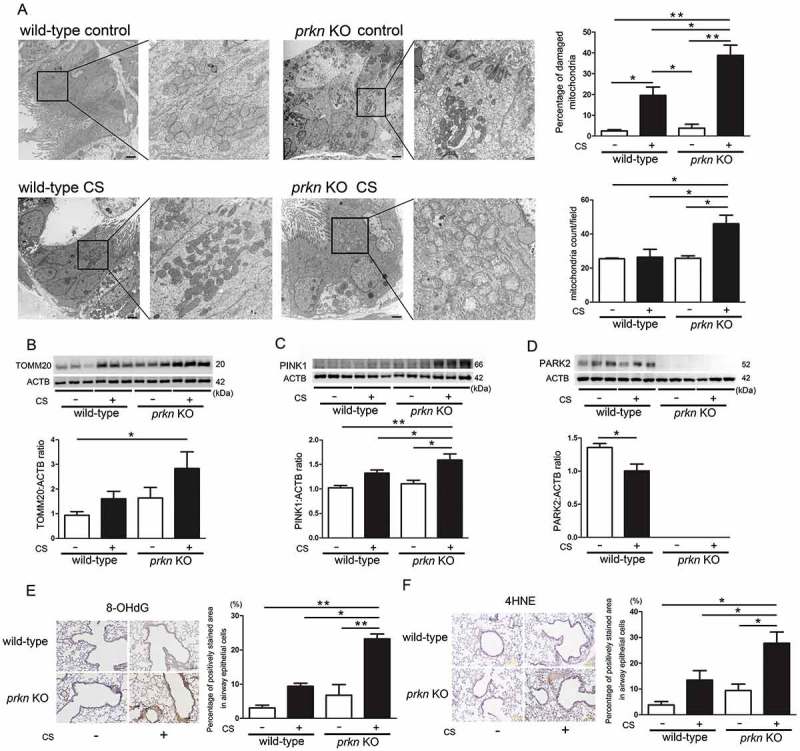
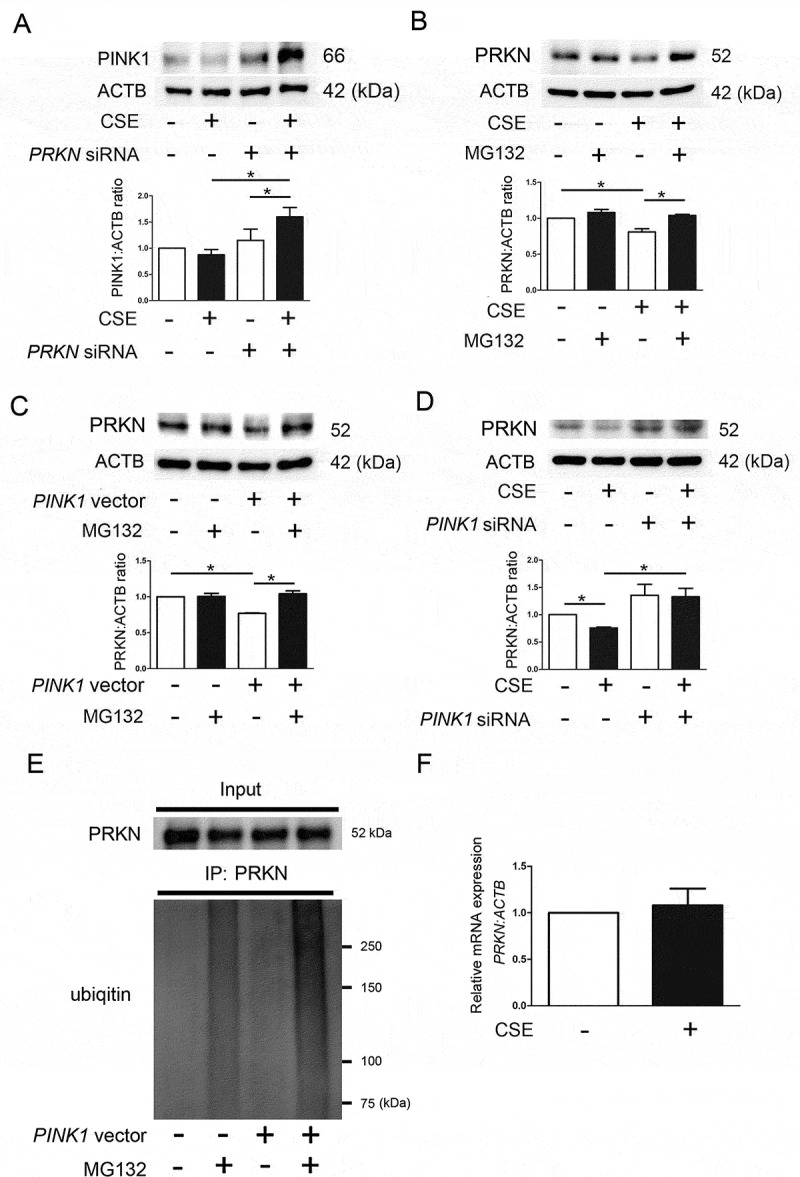
Cigarette smoke (CS)-induced accumulation of mitochondrial damage has been widely implicated in chronic obstructive pulmonary disease (COPD) pathogenesis. Mitophagy plays a crucial role in eliminating damaged mitochondria, and is governed by the PINK1 (PTEN induced putative protein kinase 1)-PRKN (parkin RBR E3 ubiquitin protein ligase) pathway. Although both increased PINK1 and reduced PRKN have been implicated in COPD pathogenesis in association with mitophagy, there are conflicting reports for the role of mitophagy in COPD progression. To clarify the involvement of PRKN-regulated mitophagy in COPD pathogenesis, prkn knockout (KO) mouse models were used. To illuminate how PINK1 and PRKN regulate mitophagy in relation to CS-induced mitochondrial damage and cellular senescence, overexpression and knockdown experiments were performed in airway epithelial cells (AEC). In comparison to wild-type mice, prkn KO mice demonstrated enhanced airway wall thickening with emphysematous changes following CS exposure. AEC in CS-exposed prkn KO mice showed accumulation of damaged mitochondria and increased oxidative modifications accompanied by accelerated cellular senescence. In vitro experiments showed PRKN overexpression was sufficient to induce mitophagy during CSE exposure even in the setting of reduced PINK1 protein levels, resulting in attenuation of mitochondrial ROS production and cellular senescence. Conversely PINK1 overexpression failed to recover impaired mitophagy caused by PRKN knockdown, indicating that PRKN protein levels can be the rate-limiting factor in PINK1-PRKN-mediated mitophagy during CSE exposure. These results suggest that PRKN levels may play a pivotal role in COPD pathogenesis by regulating mitophagy, suggesting that PRKN induction could mitigate the progression of COPD. Abbreviations: AD: Alzheimer disease; AEC: airway epithelial cells; BALF: bronchoalveolar lavage fluid; AKT: AKT serine/threonine kinase; CALCOCO2/NDP52: calcium binding and coiled-coil domain 2; CDKN1A: cyclin dependent kinase inhibitor 1A; CDKN2A: cyclin dependent kinase inhibitor 2A; COPD: chronic obstructive pulmonary disease; CS: cigarette smoke; CSE: CS extract; CXCL1: C-X-C motif chemokine ligand 1; CXCL8: C-X-C motif chemokine ligand 8; HBEC: human bronchial epithelial cells; 4-HNE: 4-hydroxynonenal; IL: interleukin; KO: knockout; LF: lung fibroblasts; LPS: lipopolysaccharide; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MTOR: mechanistic target of rapamycin kinase; 8-OHdG: 8-hydroxy-2'-deoxyguanosine; OPTN: optineurin; PRKN: parkin RBR E3 ubiquitin protein ligase; PCD: programmed cell death; PFD: pirfenidone; PIK3C: phosphatidylinositol-4:5-bisphosphate 3-kinase catalytic subunit; PINK1: PTEN induced putative kinase 1; PTEN: phosphatase and tensin homolog; RA: rheumatoid arthritis; ROS: reactive oxygen species; SA-GLB1/β-Gal: senescence-associated-galactosidase, beta 1; SASP: senescence-associated secretory phenotype; SNP: single nucleotide polymorphism; TNF: tumor necrosis factor.
Keywords: COPD; Cellular senescence; PINK1; PRKN; mitophagy.
Figures
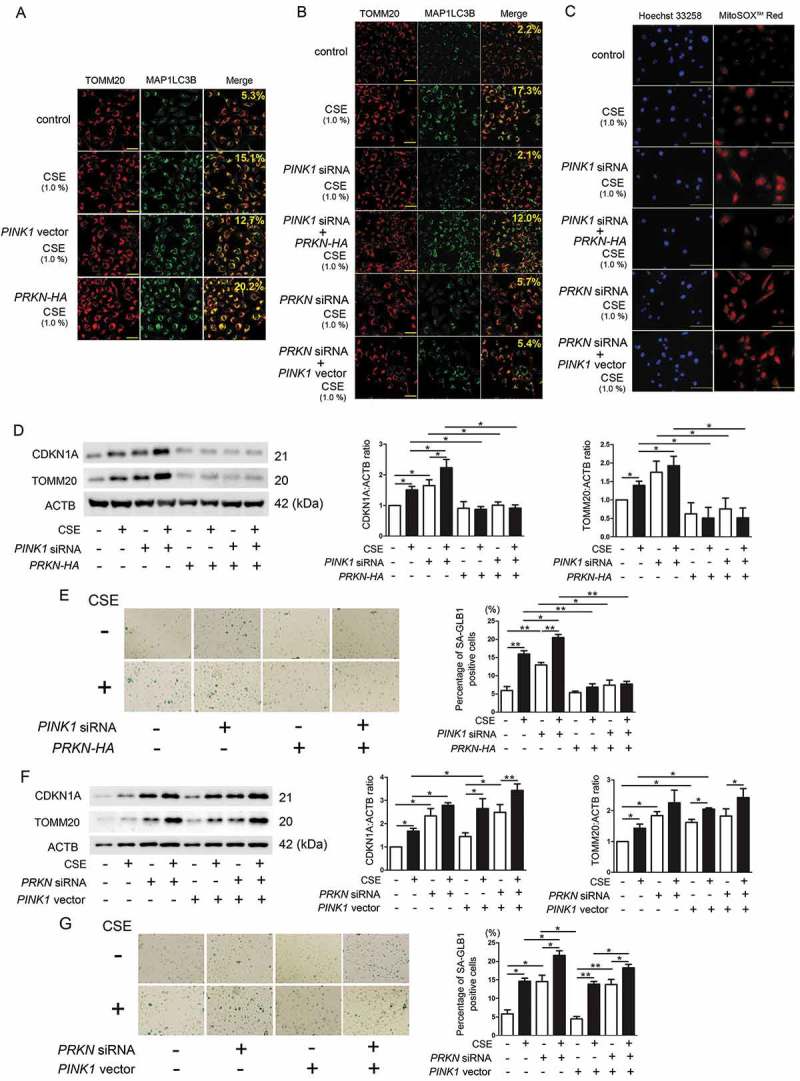
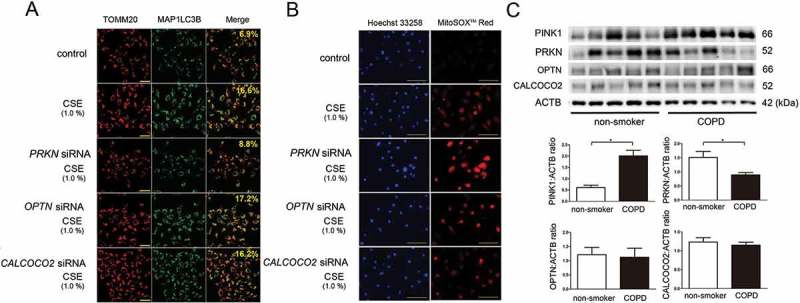
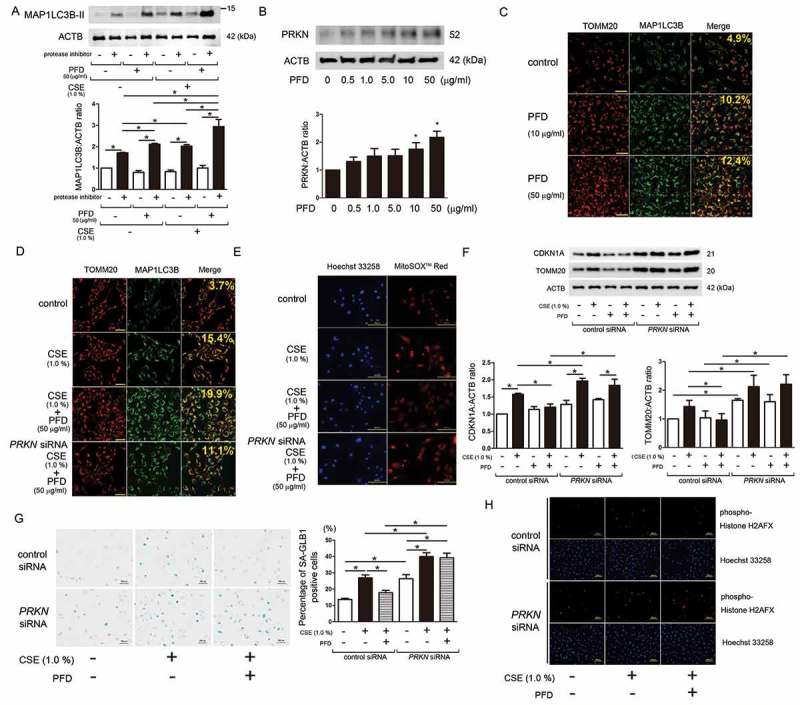
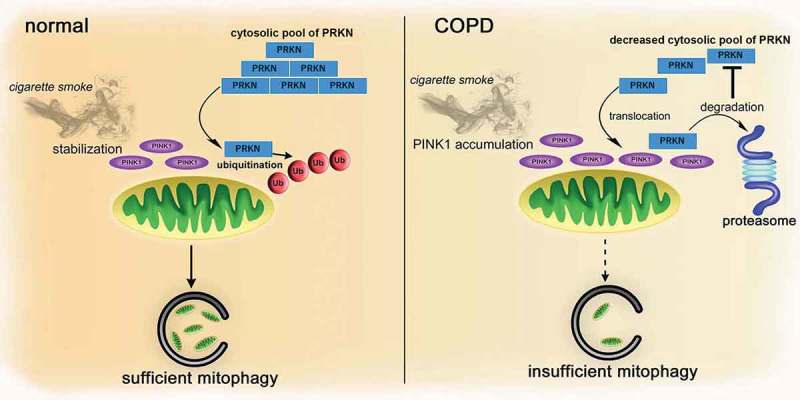
References
-
- van der Toorn M, Slebos DJ, de Bruin HG, et al. Cigarette smoke-induced blockade of the mitochondrial respiratory chain switches lung epithelial cell apoptosis into necrosis. Am J Physiol Lung Cell Mol Physiol. 2007. May;292(5):L1211–218. PubMed PMID: 17209140. - PubMed
-
- van der Toorn M, Rezayat D, Kauffman HF, et al. Lipid-soluble components in cigarette smoke induce mitochondrial production of reactive oxygen species in lung epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2009. July;297(1):L109–114. PubMed PMID: 19411310; PubMed Central PMCID: PMCPMC2711811. - PMC - PubMed
-
- Hara H, Araya J, Ito S, et al. Mitochondrial fragmentation in cigarette smoke-induced bronchial epithelial cell senescence. Am J Physiol Lung Cell Mol Physiol. 2013. November 15;305(10):L737–746. PubMed PMID: 24056969. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous