

Genome-wide association scan for heterotic quantitative trait loci in multi-breed and crossbred beef cattle
- PMID: 30290764
- PMCID: PMC6173862
- DOI: 10.1186/s12711-018-0405-y
Genome-wide association scan for heterotic quantitative trait loci in multi-breed and crossbred beef cattle
Abstract
Background: Heterosis has been suggested to be caused by dominance effects. We performed a joint genome-wide association analysis (GWAS) using data from multi-breed and crossbred beef cattle to identify single nucleotide polymorphisms (SNPs) with significant dominance effects associated with variation in growth and carcass traits and to understand the mode of action of these associations.
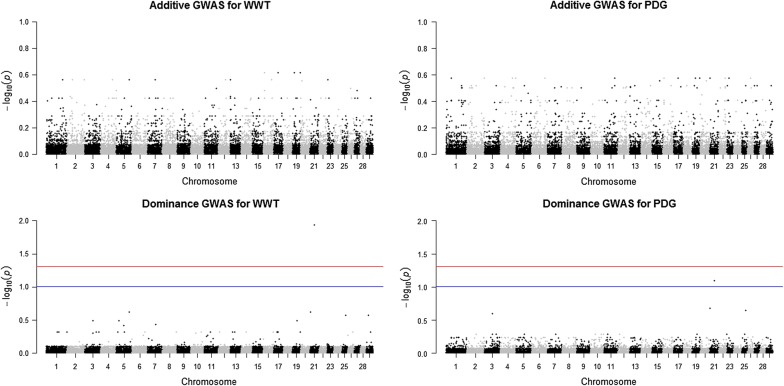
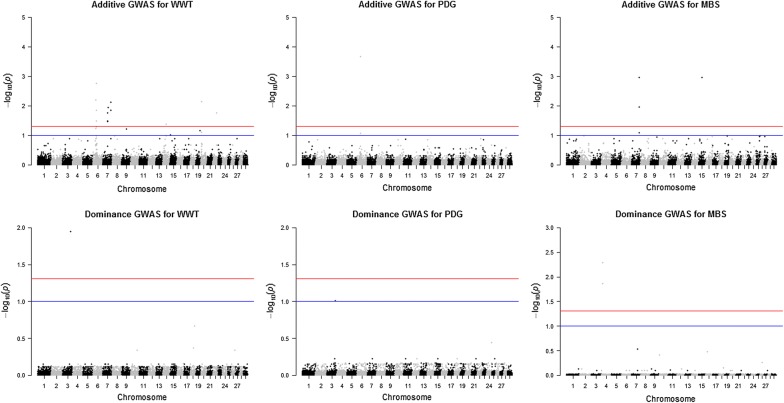
Methods: Illumina BovineSNP50 genotypes and phenotypes for 11 growth and carcass traits were available for 6796 multi-breed and crossbred beef cattle. After performing quality control, 42,610 SNPs and 6794 animals were used for further analyses. A single-SNP GWAS for the joint association of additive and dominance effects was conducted in purebred, crossbred, and combined datasets using the ASReml software. Genomic breed composition predicted from admixture analyses was included in the mixed effect model to account for possible population stratification and breed effects. A threshold of 10% genome-wide false discovery rate was applied to declare associations as significant. The significant SNPs with dominance association were mapped to their corresponding genes at 100 kb.
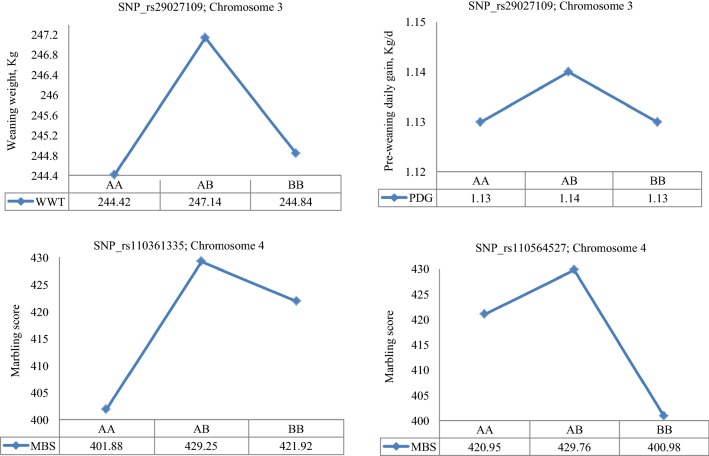
Results: Seven SNPs with significant dominance associations were detected for birth weight, weaning weight, pre-weaning daily gain, yearling weight and marbling score across the three datasets at a false discovery rate of 10%. These SNPs were located on bovine chromosomes 1, 3, 4, 6 and 21 and mapped to six putative candidate genes: U6atac, AGBL4, bta-mir-2888-1, REPIN1, ICA1 and NXPH1. These genes have interesting biological functions related to the regulation of gene expression, glucose and lipid metabolism and body fat mass. For most of the identified loci, we observed over-dominance association with the studied traits, such that the heterozygous individuals at any of these loci had greater genotypic values for the trait than either of the homozygous individuals.
Conclusions: Our results revealed very few regions with significant dominance genetic effects across all the traits studied in the three datasets used. Regarding the SNPs that were detected with dominance associations, further investigation is needed to determine their relevance in crossbreeding programs assuming that dominance effects are the main cause of (or contribute usefully to) heterosis.
Figures




References
-
- Saatchi M, Beever JE, Decker JE, Faulkner DB, Freetly HC, Hansen SL, et al. QTLs associated with dry matter intake, metabolic mid-test weight, growth and feed efficiency have little overlap across 4 beef cattle studies. BMC Genomics. 2014;15:1004. doi: 10.1186/1471-2164-15-1004. - DOI - PMC - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
