

Site-Specific Labelling of Multidomain Proteins by Amber Codon Suppression
- PMID: 30291265
- PMCID: PMC6173736
- DOI: 10.1038/s41598-018-33115-5
Site-Specific Labelling of Multidomain Proteins by Amber Codon Suppression
Abstract
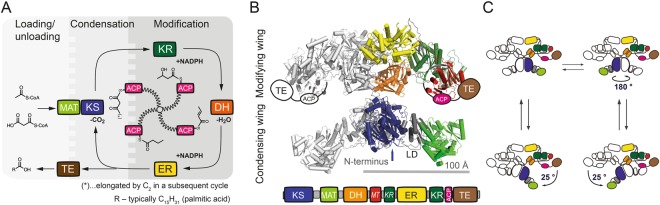
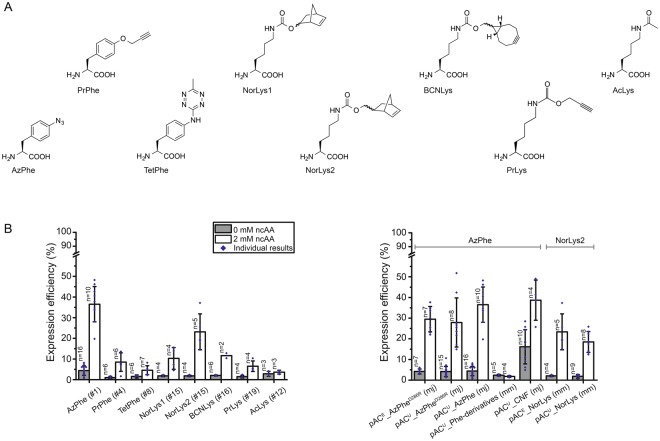
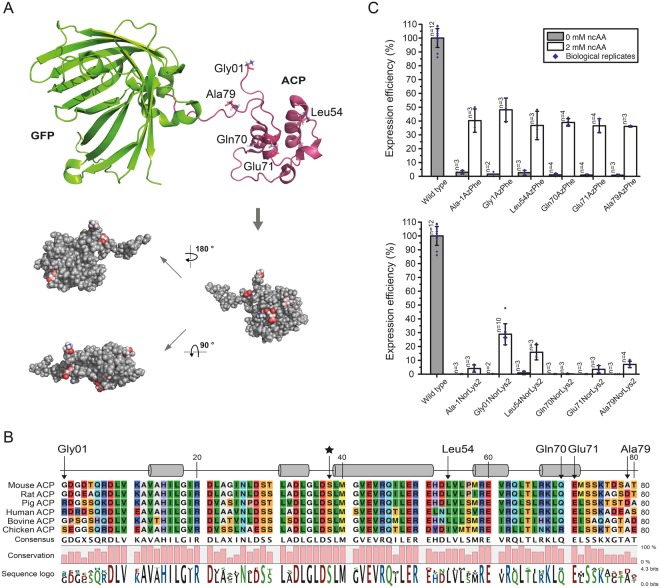
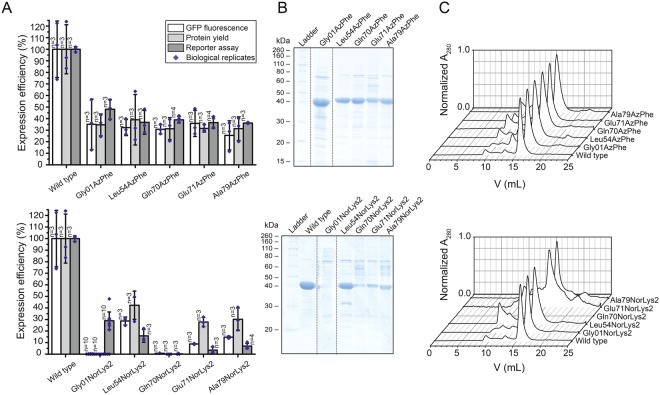
The access to information on the dynamic behaviour of large proteins is usually hindered as spectroscopic methods require the site-specific attachment of biophysical probes. A powerful emerging tool to tackle this issue is amber codon suppression. Till date, its application on large and complex multidomain proteins of MDa size has not been reported. Herein, we systematically investigate the feasibility to introduce different non-canonical amino acids into a 540 kDa homodimeric fatty acid synthase type I by genetic code expansion with subsequent fluorescent labelling. Our approach relies on a microplate-based reporter assay of low complexity using a GFP fusion protein to quickly screen for sufficient suppression conditions. Once identified, these findings were successfully utilized to upscale both the expression scale and the protein size to full-length constructs. These fluorescently labelled samples of fatty acid synthase were subjected to initial biophysical experiments, including HPLC analysis, activity assays and fluorescence spectroscopy. Successful introduction of such probes into a molecular machine such as fatty acid synthases may pave the way to understand the conformational variability, which is a primary intrinsic property required for efficient interplay of all catalytic functionalities, and to engineer them.
Conflict of interest statement
The authors declare no competing interests.
Figures
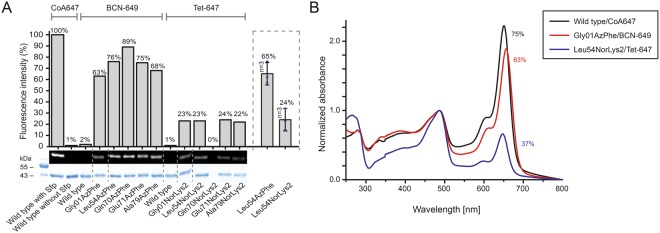
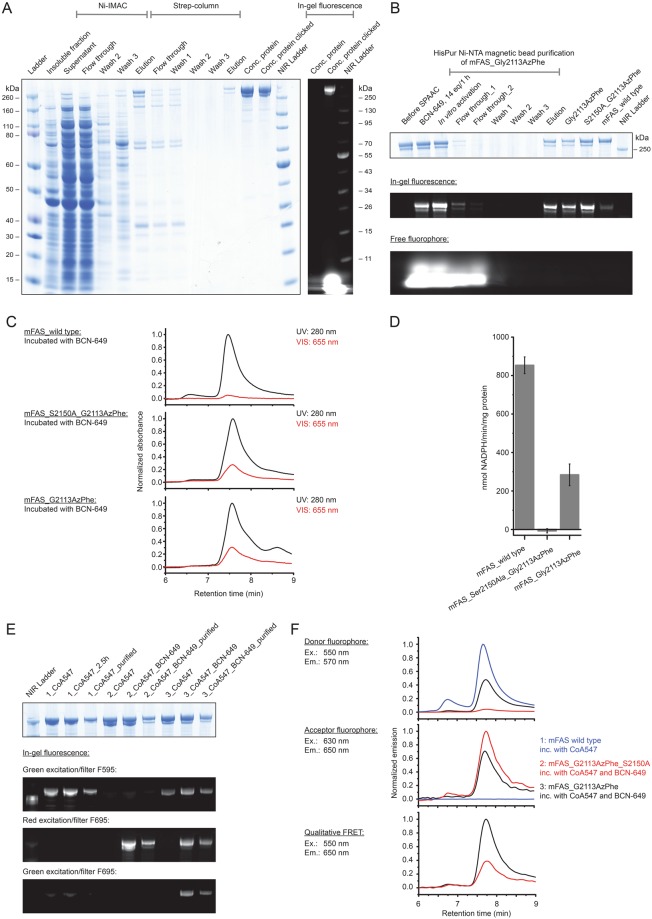
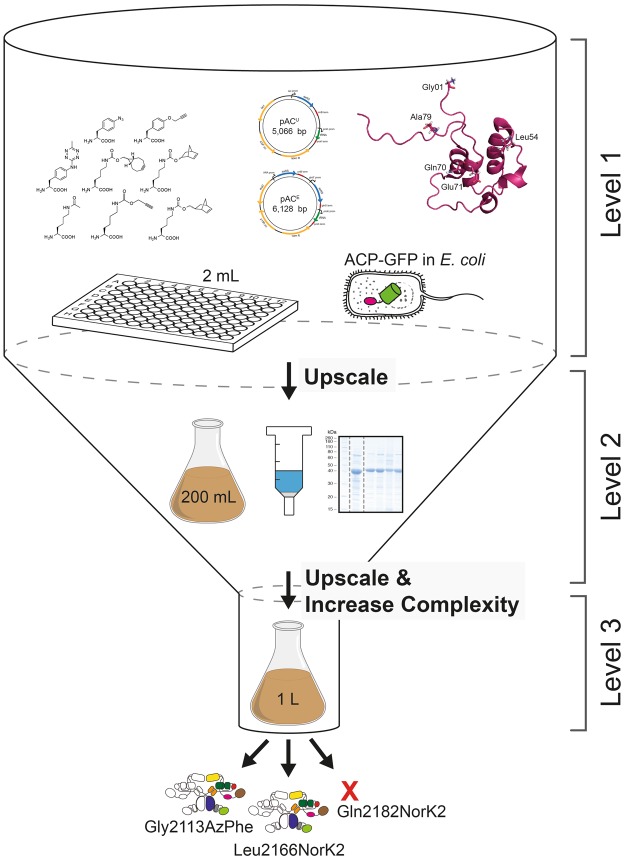
References
-
- Chang S-I, Hammes GG. Structure and Mechanism of Action of a Multifunctional Enzyme: Fatty Acid Synthase. Acc. Chem. Res. 1990;23:363–369. doi: 10.1021/ar00179a003. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
