

M1 Macrophage Polarization Is Dependent on TRPC1-Mediated Calcium Entry
- PMID: 30293012
- PMCID: PMC6174824
- DOI: 10.1016/j.isci.2018.09.014
M1 Macrophage Polarization Is Dependent on TRPC1-Mediated Calcium Entry
Abstract
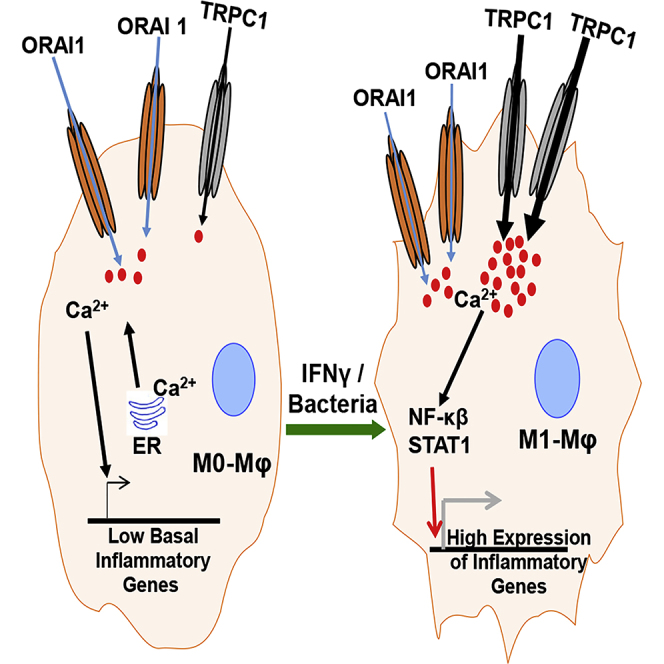
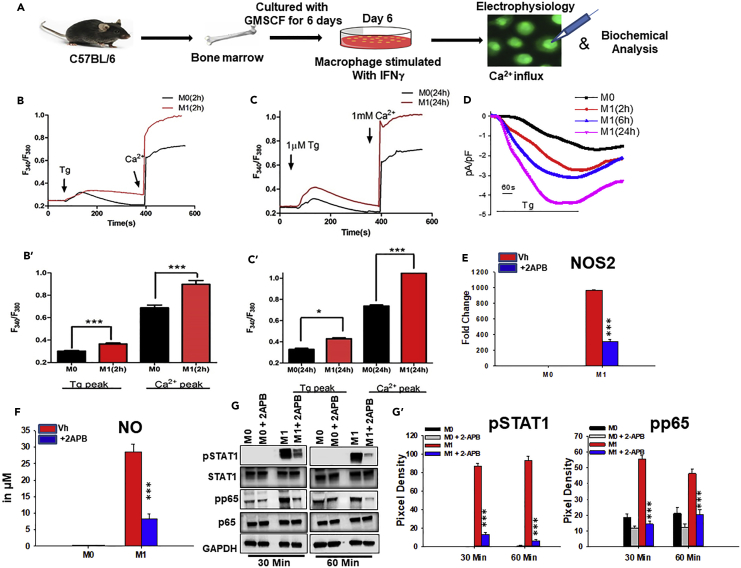
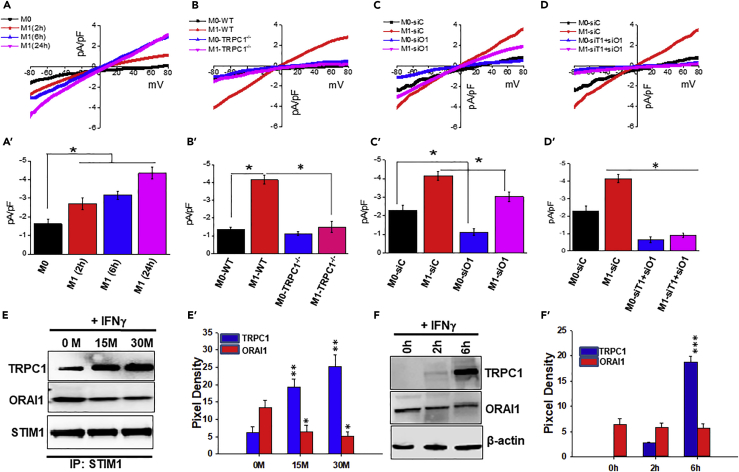
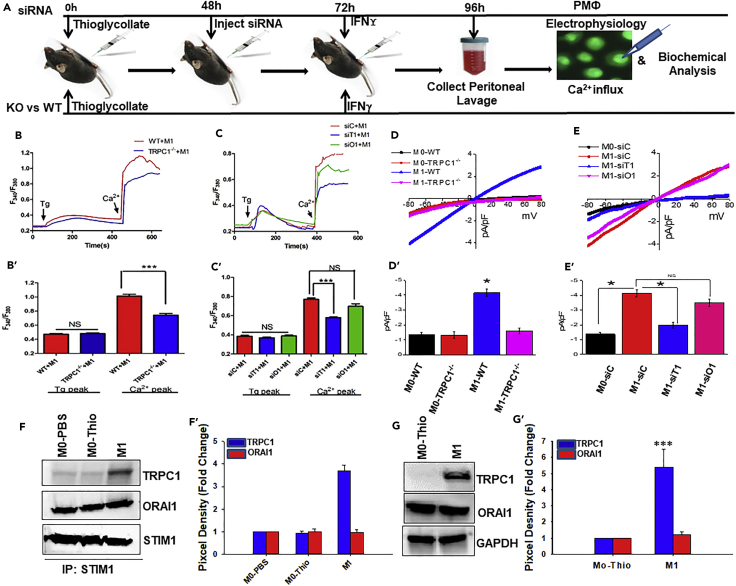
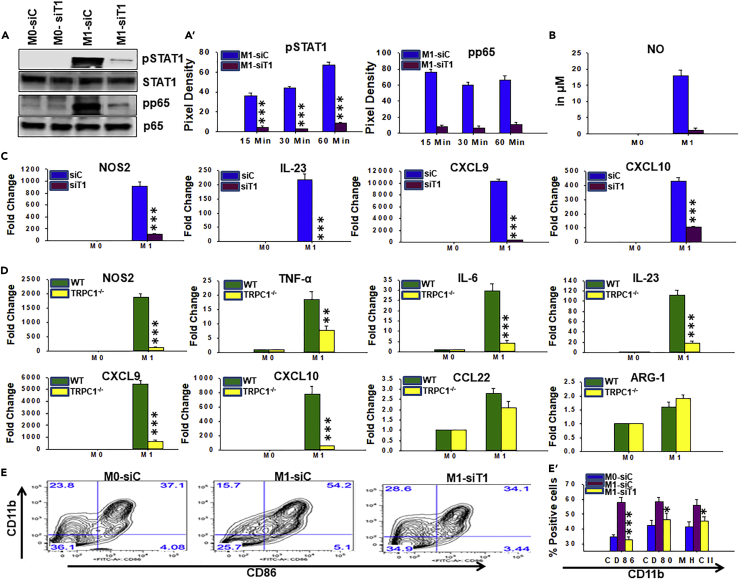
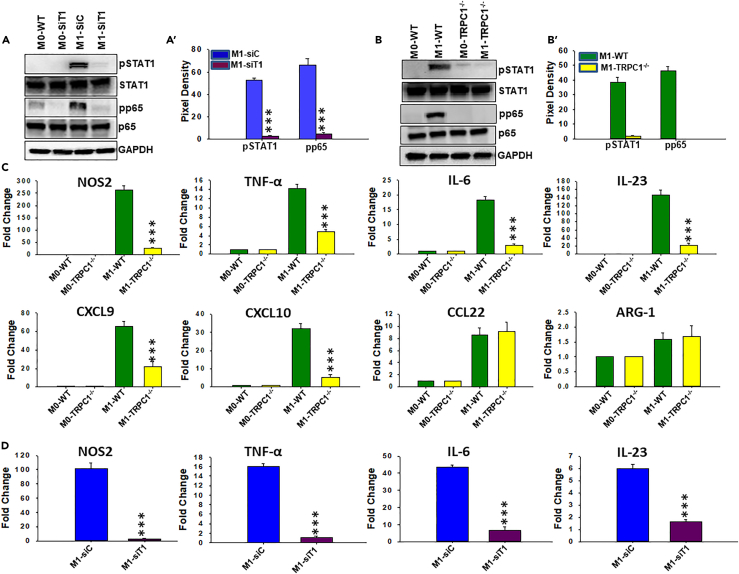
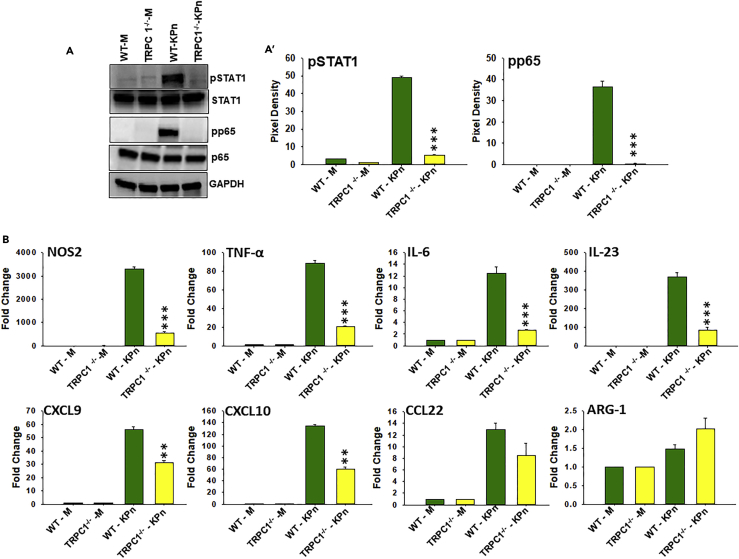
Macrophage plasticity is essential for innate immunity, but in-depth signaling mechanism(s) regulating their functional phenotypes are ill-defined. Here we report that interferon (IFN) γ priming of naive macrophages induces store-mediated Ca2+ entry and inhibition of Ca2+ entry impairs polarization to M1 inflammatory phenotype. In vitro and in vivo functional analyses revealed ORAI1 to be a primary contributor to basal Ca2+ influx in macrophages, whereas IFNγ-induced Ca2+ influx was mediated by TRPC1. Deficiency of TRPC1 displayed abrogated IFNγ-induced M1 inflammatory mediators in macrophages. In a preclinical model of peritonitis by Klebsiella pneumoniae infection, macrophages showed increased Ca2+ influx, which was TRPC1 dependent. Macrophages from infected TRPC1-/- mice showed inhibited expression of M1-associated signature molecules. Furthermore, in human patients with systemic inflammatory response syndrome, the level of TRPC1 expression in circulating macrophages directly correlated with M1 inflammatory mediators. Overall, TRPC1-mediated Ca2+ influx is essential for the induction/shaping of macrophage polarization to M1 inflammatory phenotype.
Keywords: Biological Sciences; Immune Response; Immunology.
Copyright © 2018 The Author(s). Published by Elsevier Inc. All rights reserved.
Figures

References
-
- Braun A., Gessner J.E., Varga-Szabo D., Syed S.N., Konrad S., Stegner D., Vogtle T., Schmidt R.E., Nieswandt B. STIM1 is essential for Fcgamma receptor activation and autoimmune inflammation. Blood. 2009;113:1097–1104. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
