

The first pediatric case of glucagon receptor defect due to biallelic mutations in GCGR is identified by newborn screening of elevated arginine
- PMID: 30294546
- PMCID: PMC6171159
- DOI: 10.1016/j.ymgmr.2018.09.006
The first pediatric case of glucagon receptor defect due to biallelic mutations in GCGR is identified by newborn screening of elevated arginine
Abstract
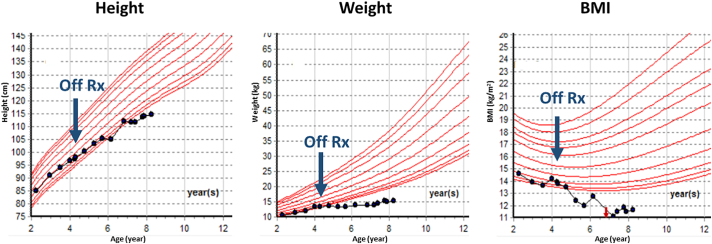
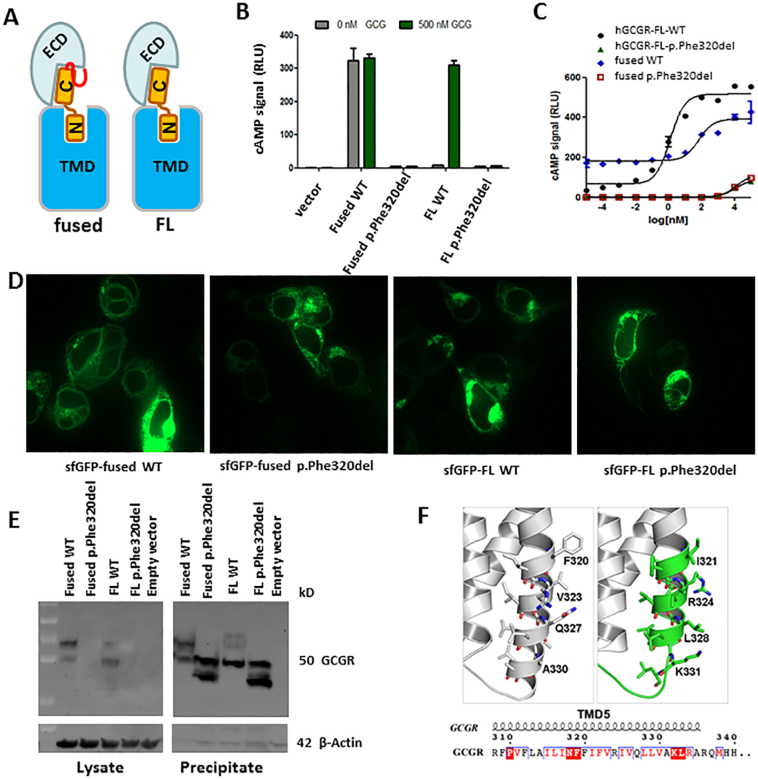
Glucagon receptor (GCGR) defect (Mahvash disease) is an autosomal recessive hereditary pancreatic neuroendocrine tumor (PNET) syndrome that has only been reported in adults with pancreatic α cell hyperplasia and PNETs. We describe a 7-year-old girl with persistent hyperaminoacidemia, notable for elevations of glutamine (normal ammonia), alanine (normal lactate), dibasic amino acids (arginine, lysine and ornithine), threonine and serine. She initially was brought to medical attention by an elevated arginine on newborn screening (NBS) and treated for presumed arginase deficiency with a low protein diet, essential amino acids formula and an ammonia scavenger drug. This treatment normalized plasma amino acids. She had intermittent emesis and anorexia, but was intellectually normal. Arginase enzyme assay and ARG1 sequencing and deletion/duplication analysis were normal. Treatments were stopped, but similar pattern of hyperaminoacidemia recurred. She also had hypercholesterolemia type IIa, with only elevated LDL cholesterol, despite an extremely lean body habitus. Exome sequencing was initially non-diagnostic. Through a literature search, we recognized the pattern of hyperaminoacidemia was strikingly similar to that reported in the Gcgr -/- knockout mice. Subsequently the patient was found to have an extremely elevated plasma glucagon and a novel, homozygous c.958_960del (p.Phe320del) variant in GCGR. Functional studies confirmed the pathogenicity of this variant. This case expands the clinical phenotype of GCGR defect in children and emphasizes the clinical utility of plasma amino acids in screening, diagnosis and monitoring glucagon signaling interruption. Early identification of a GCGR defect may provide an opportunity for potential beneficial treatment for an adult onset tumor predisposition disease.
Keywords: GCGR mutation; Glucagon receptor; Hyperaminoacidemia; Mahvash disease; Newborn screening; Pancreatic neuroendocrine tumor (PNET); Pancreatic α cell hyperplasia (ACH).
Figures



Similar articles
-
Absence of PNET formation and normal longevity in a mouse model of Mahvash disease.Heliyon. 2024 Jul 26;10(15):e35362. doi: 10.1016/j.heliyon.2024.e35362. eCollection 2024 Aug 15. Heliyon. 2024. PMID: 39170309 Free PMC article.
-
Familial Mahvash disease with metastatic pancreatic NET and MEN1 mutations.Endocr Relat Cancer. 2025 Jun 9;32(6):e250087. doi: 10.1530/ERC-25-0087. Print 2025 Jun 1. Endocr Relat Cancer. 2025. PMID: 40424057
-
Glucagon receptor is required for long-term survival: a natural history study of the Mahvash disease in a murine model.Endocrinol Nutr. 2012 Nov;59(9):523-30. doi: 10.1016/j.endonu.2012.06.006. Epub 2012 Aug 28. Endocrinol Nutr. 2012. PMID: 22951296
-
Pancreatic α-cell hyperplasia: facts and myths.J Clin Endocrinol Metab. 2014 Mar;99(3):748-56. doi: 10.1210/jc.2013-2952. Epub 2013 Nov 27. J Clin Endocrinol Metab. 2014. PMID: 24285676 Review.
-
Glucagon cell hyperplasia and neoplasia: a recently recognized endocrine receptor disease.Endocr Relat Cancer. 2023 Jul 18;30(8):e230032. doi: 10.1530/ERC-23-0032. Print 2023 Aug 1. Endocr Relat Cancer. 2023. PMID: 37260318 Review.
Cited by
-
Newly discovered endocrine functions of the liver.World J Hepatol. 2021 Nov 27;13(11):1611-1628. doi: 10.4254/wjh.v13.i11.1611. World J Hepatol. 2021. PMID: 34904032 Free PMC article. Review.
-
The Role of the α Cell in the Pathogenesis of Diabetes: A World beyond the Mirror.Int J Mol Sci. 2021 Sep 1;22(17):9504. doi: 10.3390/ijms22179504. Int J Mol Sci. 2021. PMID: 34502413 Free PMC article. Review.
-
Inborn Errors of Metabolism in the Era of Untargeted Metabolomics and Lipidomics.Metabolites. 2019 Oct 21;9(10):242. doi: 10.3390/metabo9100242. Metabolites. 2019. PMID: 31640247 Free PMC article. Review.
-
Late gestation fetal hyperglucagonaemia impairs placental function and results in diminished fetal protein accretion and decreased fetal growth.J Physiol. 2021 Jul;599(13):3403-3427. doi: 10.1113/JP281288. Epub 2021 May 10. J Physiol. 2021. PMID: 33878802 Free PMC article.
-
Proprotein convertase subtilisin/kexin type 9 and lipid metabolism.Curr Opin Lipidol. 2019 Jun;30(3):186-191. doi: 10.1097/MOL.0000000000000601. Curr Opin Lipidol. 2019. PMID: 30925519 Free PMC article. Review.
References
-
- Yu R., Nissen N.N., Dhall D., Heaney A.P. Nesidioblastosis and hyperplasia of alpha cells, microglucagonoma, and nonfunctioning islet cell tumor of the pancreas: review of the literature. Pancreas. 2008;36(4):428–431. - PubMed
-
- Yu R. Mahvash Disease: 10 Years After Discovery. Pancreas. 2018;47(5):511–515. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
