

N-acetylcysteine targets 5 lipoxygenase-derived, toxic lipids and can synergize with prostaglandin E2 to inhibit ferroptosis and improve outcomes following hemorrhagic stroke in mice
- PMID: 30294906
- PMCID: PMC6519209
- DOI: 10.1002/ana.25356
N-acetylcysteine targets 5 lipoxygenase-derived, toxic lipids and can synergize with prostaglandin E2 to inhibit ferroptosis and improve outcomes following hemorrhagic stroke in mice
Abstract
Objectives: N-acetylcysteine (NAC) is a clinically approved thiol-containing redox modulatory compound currently in trials for many neurological and psychiatric disorders. Although generically labeled as an "antioxidant," poor understanding of its site(s) of action is a barrier to its use in neurological practice. Here, we examined the efficacy and mechanism of action of NAC in rodent models of hemorrhagic stroke.
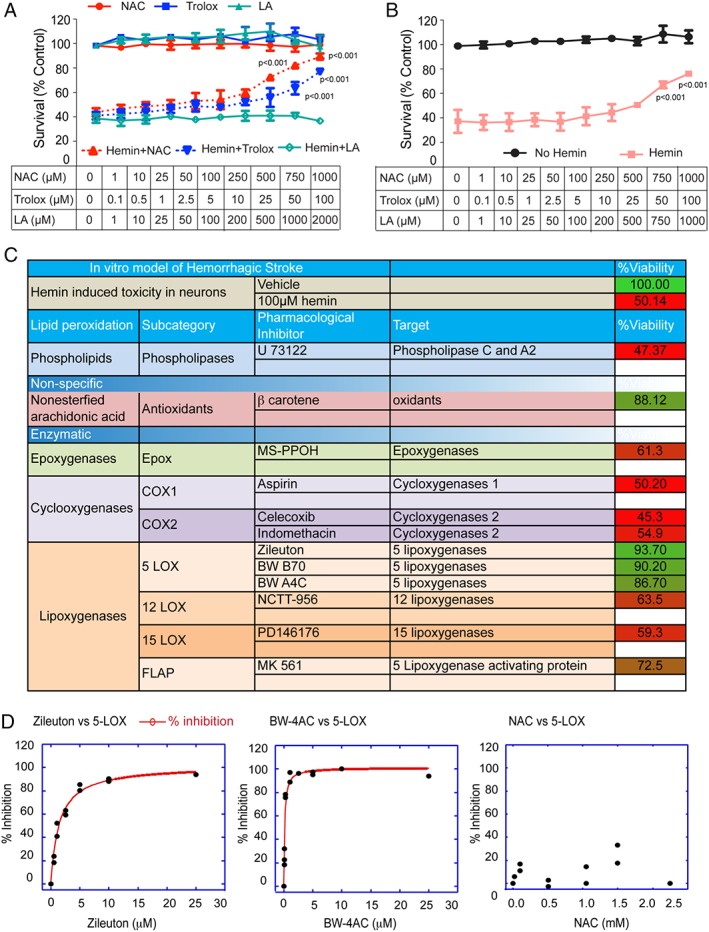
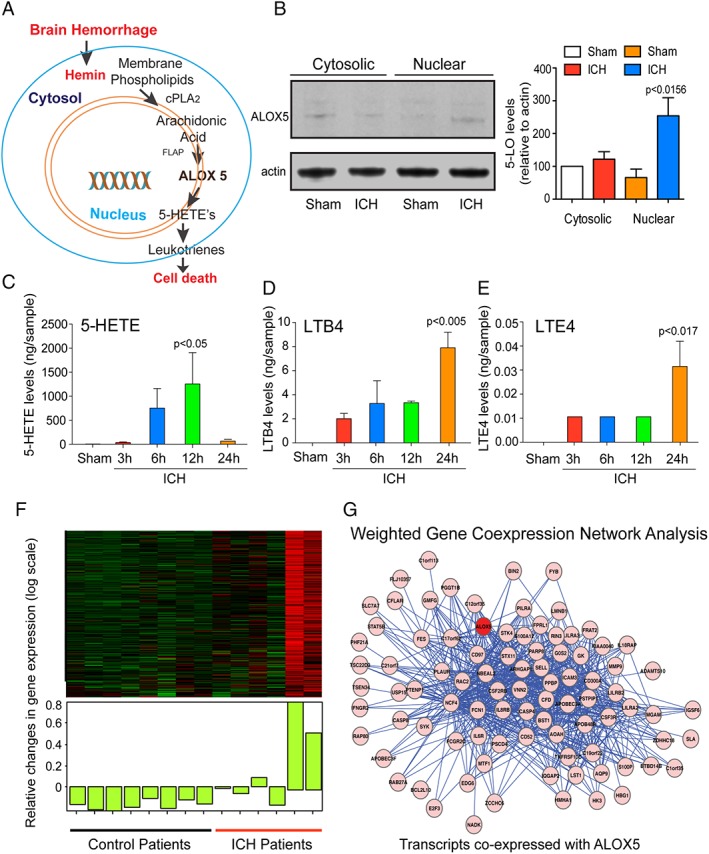
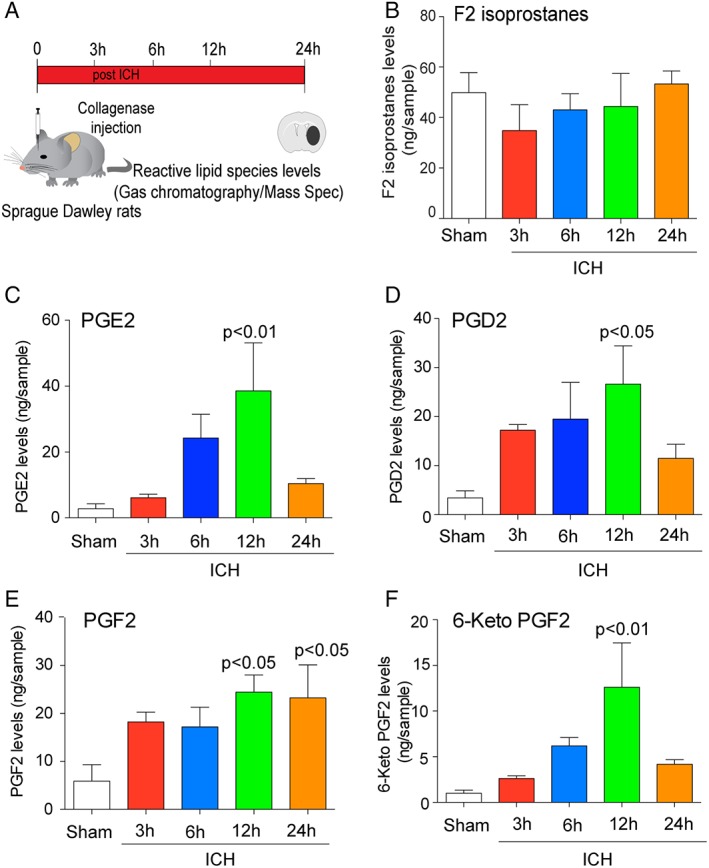
Methods: Hemin was used to model ferroptosis and hemorrhagic stroke in cultured neurons. Striatal infusion of collagenase was used to model intracerebral hemorrhage (ICH) in mice and rats. Chemical biology, targeted lipidomics, arachidonate 5-lipoxygenase (ALOX5) knockout mice, and viral-gene transfer were used to gain insight into the pharmacological targets and mechanism of action of NAC.
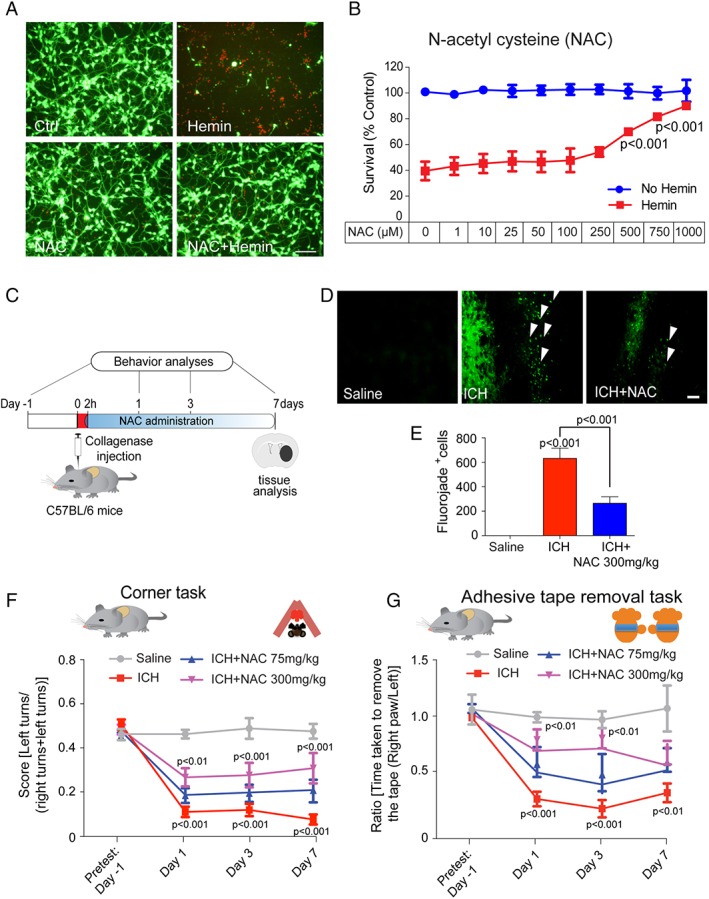
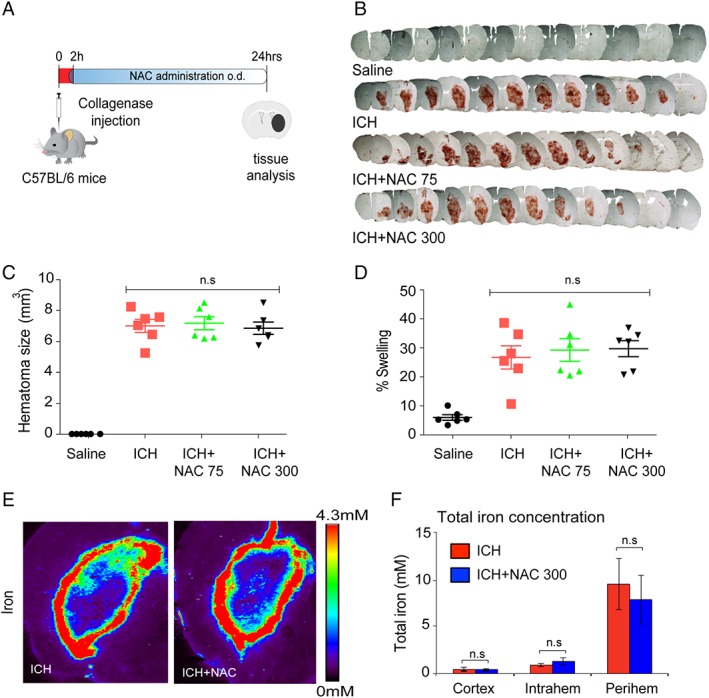
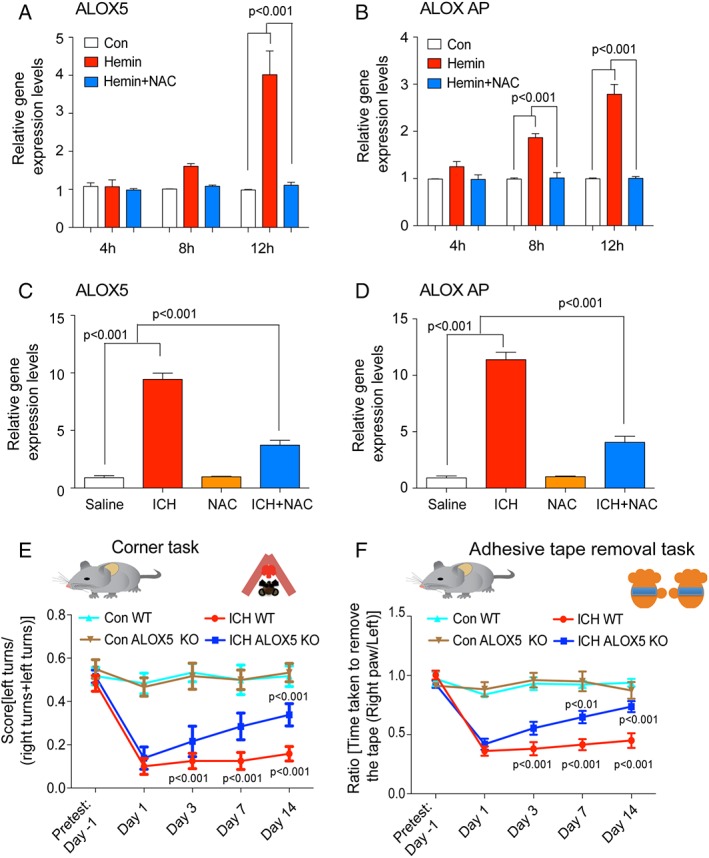
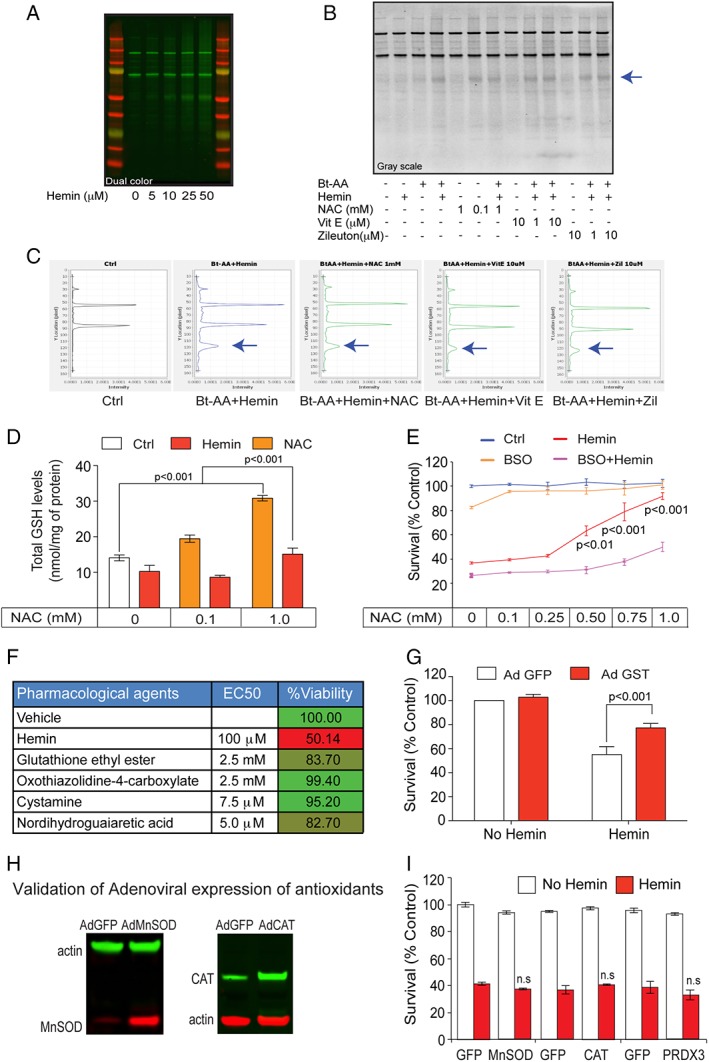
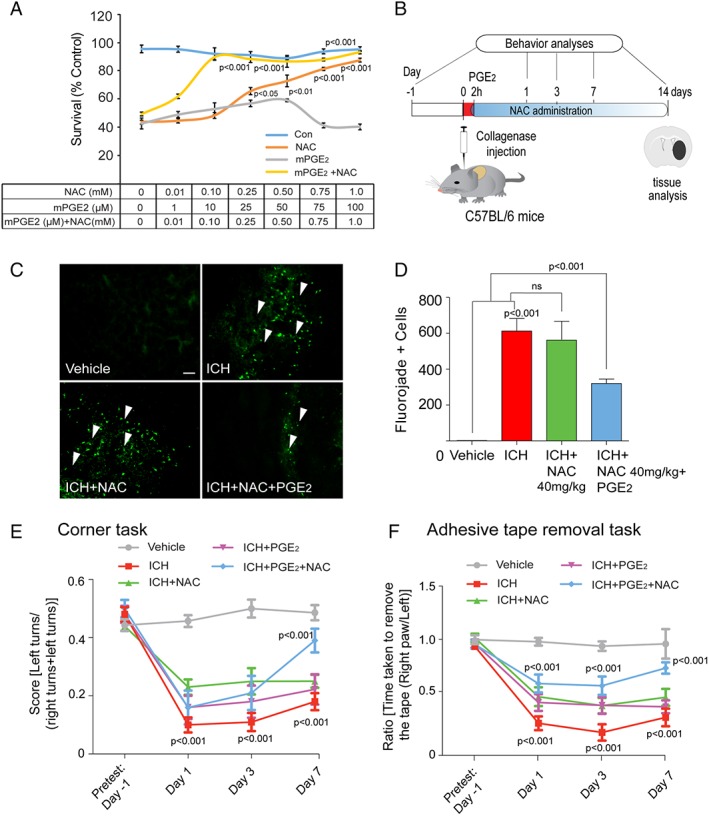
Results: NAC prevented hemin-induced ferroptosis by neutralizing toxic lipids generated by arachidonate-dependent ALOX5 activity. NAC efficacy required increases in glutathione and is correlated with suppression of reactive lipids by glutathione-dependent enzymes such as glutathione S-transferase. Accordingly, its protective effects were mimicked by chemical or molecular lipid peroxidation inhibitors. NAC delivered postinjury reduced neuronal death and improved functional recovery at least 7 days following ICH in mice and can synergize with clinically approved prostaglandin E2 (PGE2 ).
Interpretation: NAC is a promising, protective therapy for ICH, which acted to inhibit toxic arachidonic acid products of nuclear ALOX5 that synergized with exogenously delivered protective PGE2 in vitro and in vivo. The findings provide novel insight into a target for NAC, beyond the generic characterization as an antioxidant, resulting in neuroprotection and offer a feasible combinatorial strategy to optimize efficacy and safety in dosing of NAC for treatment of neurological disorders involving ferroptosis such as ICH. Ann Neurol 2018;84:854-872.
© 2018 The Authors. Annals of Neurology published by Wiley Periodicals, Inc. on behalf of American Neurological Association.
Conflict of interest statement
R.R.R. and S.S.K. are co‐inventors on a patent related to the use of N‐acetylcysteine and/or PGE2 in neurological disorders. These patents have been licensed by Neuronasal, Inc., an early‐stage biotechnology company. R.R.R. is on the SAB for Neuronasal and has a small equity interest in the company along with receiving occasional consulting fees.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
