

Treatments targeting inotropy
- PMID: 30295807
- PMCID: PMC7963133
- DOI: 10.1093/eurheartj/ehy600
Treatments targeting inotropy
Abstract
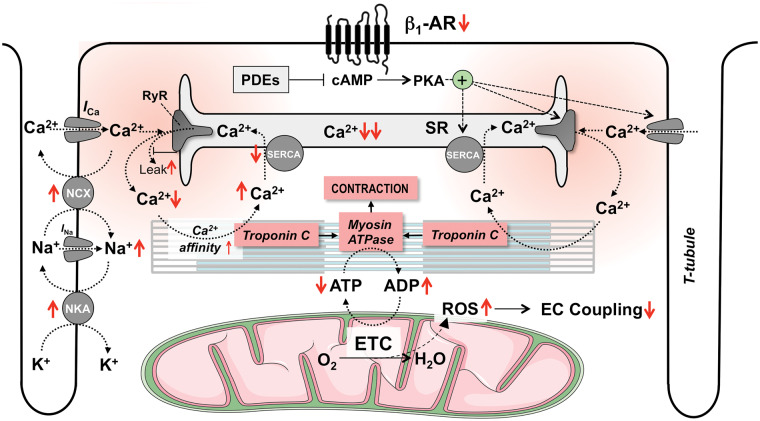
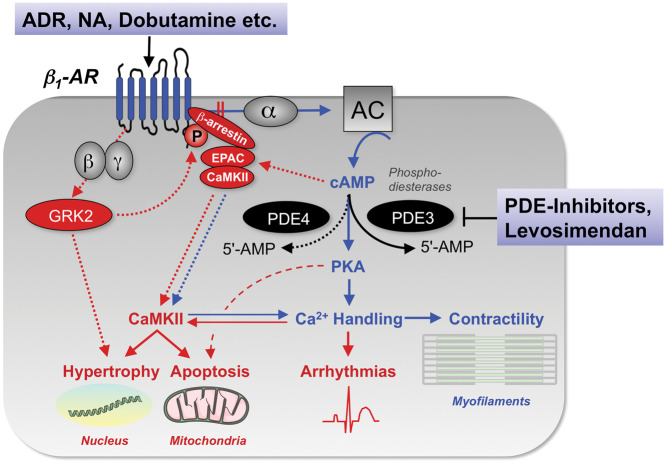
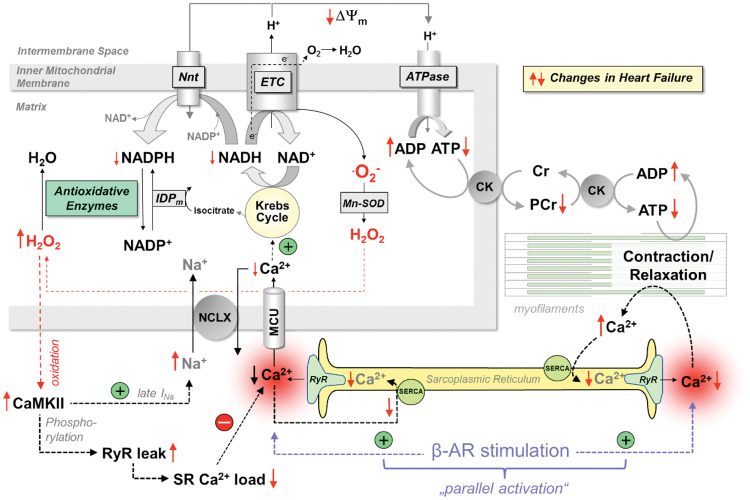
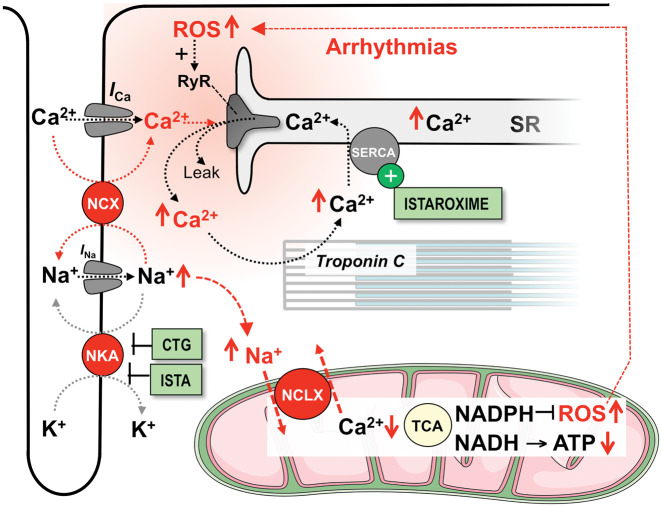
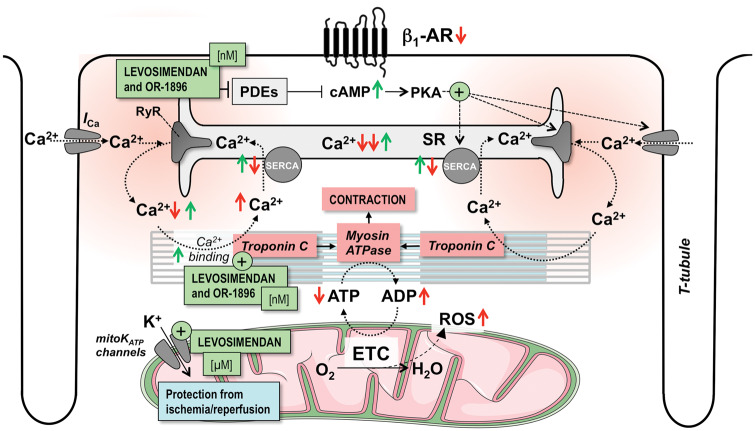
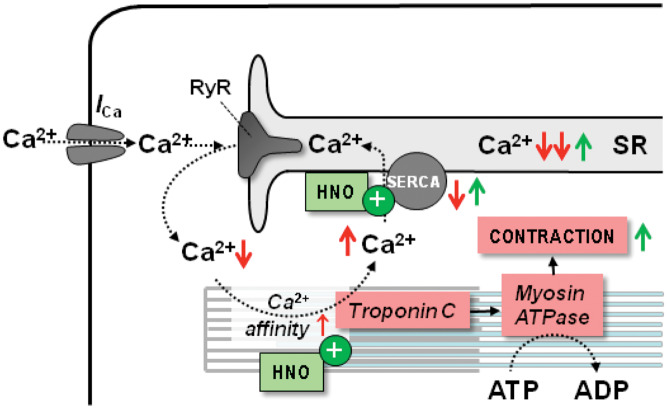
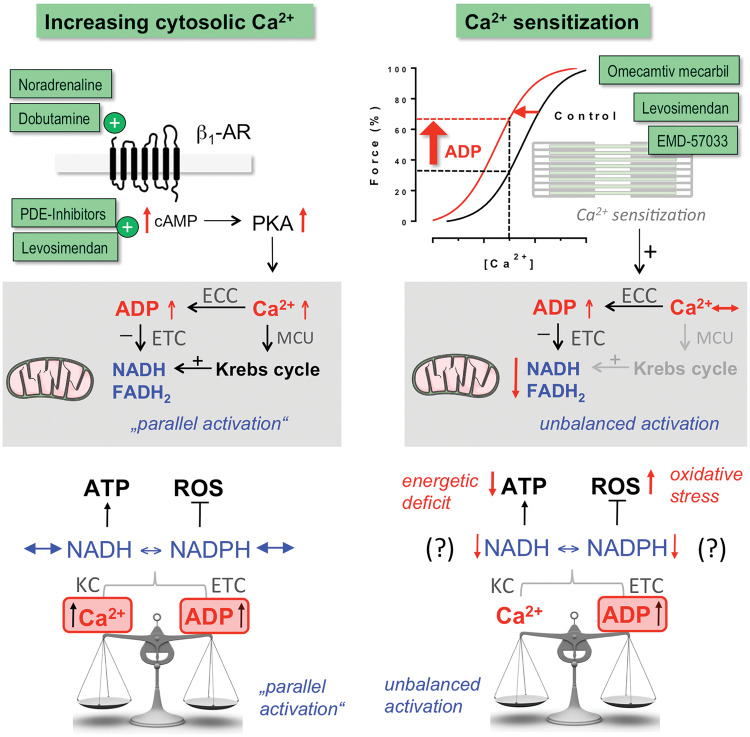
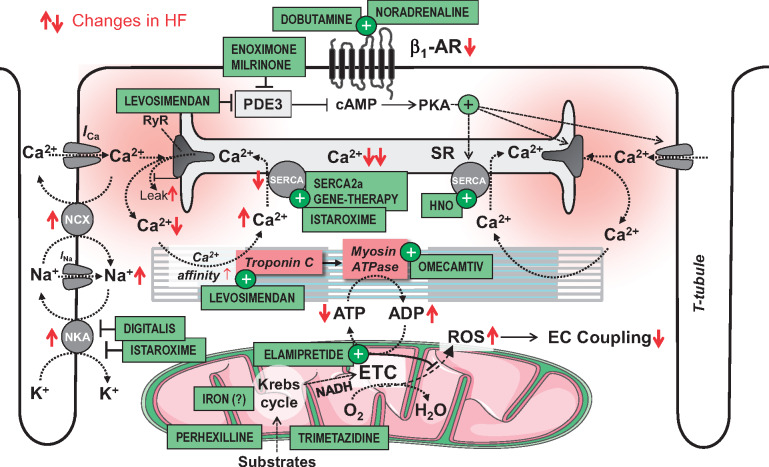
Acute heart failure (HF) and in particular, cardiogenic shock are associated with high morbidity and mortality. A therapeutic dilemma is that the use of positive inotropic agents, such as catecholamines or phosphodiesterase-inhibitors, is associated with increased mortality. Newer drugs, such as levosimendan or omecamtiv mecarbil, target sarcomeres to improve systolic function putatively without elevating intracellular Ca2+. Although meta-analyses of smaller trials suggested that levosimendan is associated with a better outcome than dobutamine, larger comparative trials failed to confirm this observation. For omecamtiv mecarbil, Phase II clinical trials suggest a favourable haemodynamic profile in patients with acute and chronic HF, and a Phase III morbidity/mortality trial in patients with chronic HF has recently begun. Here, we review the pathophysiological basis of systolic dysfunction in patients with HF and the mechanisms through which different inotropic agents improve cardiac function. Since adenosine triphosphate and reactive oxygen species production in mitochondria are intimately linked to the processes of excitation-contraction coupling, we also discuss the impact of inotropic agents on mitochondrial bioenergetics and redox regulation. Therefore, this position paper should help identify novel targets for treatments that could not only safely improve systolic and diastolic function acutely, but potentially also myocardial structure and function over a longer-term.
Keywords: Acute decompensated heart failure; Adrenergic receptors; Calcium; Cardiogenic shock; Contractility; Energetics; Excitation–contraction coupling; Heart failure; Inotropes; Levosimendan; Mitochondria; Nitroxyl; Omecamtiv mecarbil; Sarcomeres.
Published on behalf of the European Society of Cardiology. All rights reserved. © The Author(s) 2018. For permissions, please email: journals.permissions@oup.com.
Figures

References
-
- Ambrosy AP, Fonarow GC, Butler J, Chioncel O, Greene SJ, Vaduganathan M, Nodari S, Lam CSP, Sato N, Shah AN, Gheorghiade M. The global health and economic burden of hospitalizations for heart failure: lessons learned from hospitalized heart failure registries. J Am Coll Cardiol 2014;63:1123–1133. - PubMed
-
- Chioncel O, Mebazaa A, Harjola V-P, Coats AJ, Piepoli MF, Crespo-Leiro MG, Laroche C, Seferovic PM, Anker SD, Ferrari R, Ruschitzka F, Lopez-Fernandez S, Miani D, Filippatos G, Maggioni AP; on behalf of the ESC Heart Failure Long-Term Registry Investigators. Clinical phenotypes and outcome of patients hospitalized for acute heart failure: the ESC Heart Failure Long-Term Registry. Eur J Heart Fail 2017;19:1242–1254. - PubMed
-
- Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ, Falk V, Gonzalez-Juanatey JR, Harjola VP, Jankowska EA, Jessup M, Linde C, Nihoyannopoulos P, Parissis JT, Pieske B, Riley JP, Rosano GM, Ruilope LM, Ruschitzka F, Rutten FH, van der Meer P; Authors/Task Force Members; Document Reviewers. 2016 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure: the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail 2016;18:891–975. - PubMed
-
- Digitalis Investigation Group. The effect of digoxin on mortality and morbidity in patients with heart failure. N Engl J Med 1997;336:525–533. - PubMed
-
- Bers DM. Altered cardiac myocyte Ca regulation in heart failure. Physiology (Bethesda) 2006;21:380–387. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
