

Screening for Regulatory Variants in 460 kb Encompassing the CFTR Locus in Cystic Fibrosis Patients
- PMID: 30296588
- PMCID: PMC6315324
- DOI: 10.1016/j.jmoldx.2018.08.011
Screening for Regulatory Variants in 460 kb Encompassing the CFTR Locus in Cystic Fibrosis Patients
Abstract
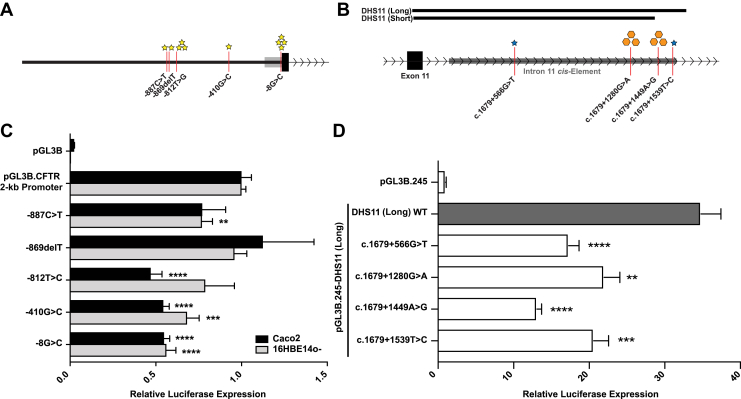
It is estimated that up to 5% of cystic fibrosis transmembrane conductance regulator (CFTR) pathogenic alleles are unidentified. Some of these errors may lie in noncoding regions of the locus and affect gene expression. To identify regulatory element variants in the CFTR locus, SureSelect targeted enrichment of 460 kb encompassing the gene was optimized to deep sequence genomic DNA from 80 CF patients with an unequivocal clinical diagnosis but only one or no CFTR-coding region pathogenic variants. Bioinformatics tools were used to identify sequence variants and predict their impact, which were then assayed in transient reporter gene luciferase assays. The effect of five variants in the CFTR promoter and four in an intestinal enhancer of the gene were assayed in relevant cell lines. The initial analysis of sequence data revealed previously known CF-causing variants, validating the robustness of the SureSelect design, and showed that 85 of 160 CF alleles were undefined. Of a total 1737 variants revealed across the extended 460-kb CFTR locus, 51 map to known CFTR cis-regulatory elements, and many of these are predicted to alter transcription factor occupancy. Four promoter variants and all those in the intestinal enhancer significantly repress reporter gene activity. These data suggest that CFTR regulatory elements may harbor novel CF disease-causing variants that warrant further investigation, both for genetic screening protocols and functional assays.
Copyright © 2019 American Society for Investigative Pathology and the Association for Molecular Pathology. Published by Elsevier Inc. All rights reserved.
Figures

References
-
- Programme WHG . 2004. The Molecular Genetic Epidemiology of Cystic Fibrosis: Report of a Joint Meeting of WHO/IECFTN/ICF(M)A/ECFS, Genoa, Italy, 19 June 2002.
-
- Massie J., Clements B., Australian Paediatric Respiratory Group Diagnosis of cystic fibrosis after newborn screening: the Australasian experience--twenty years and five million babies later: a consensus statement from the Australasian Paediatric Respiratory Group. Pediatr Pulmonol. 2005;39:440–446. - PubMed
-
- Southern K.W., Munck A., Pollitt R., Travert G., Zanolla L., Dankert-Roelse J., Castellani C., ECFS CF Neonatal Screening Working Group A survey of newborn screening for cystic fibrosis in Europe. J Cyst Fibros. 2007;6:57–65. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
