

Biosynthetic energy cost for amino acids decreases in cancer evolution
- PMID: 30297703
- PMCID: PMC6175916
- DOI: 10.1038/s41467-018-06461-1
Biosynthetic energy cost for amino acids decreases in cancer evolution
Abstract
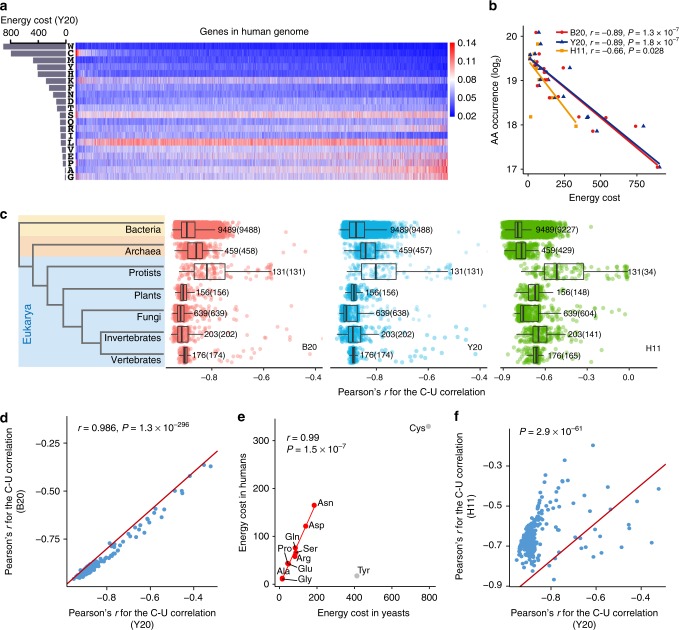
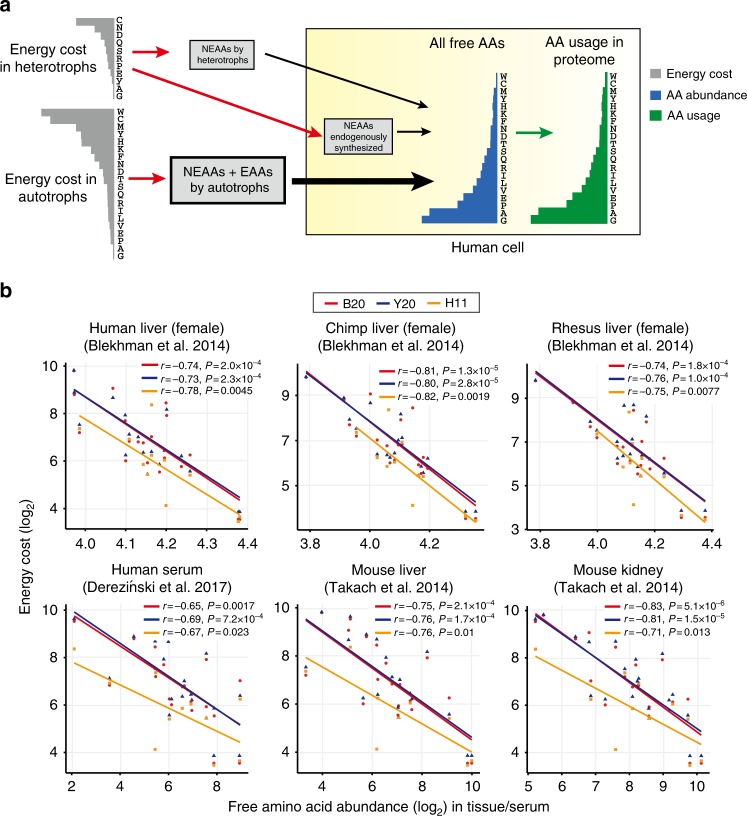
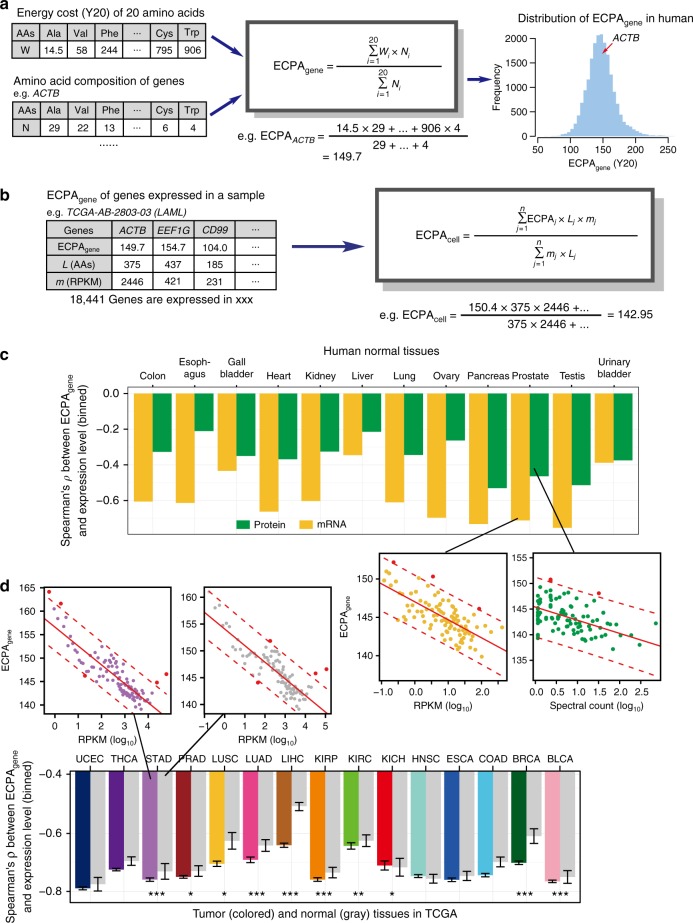
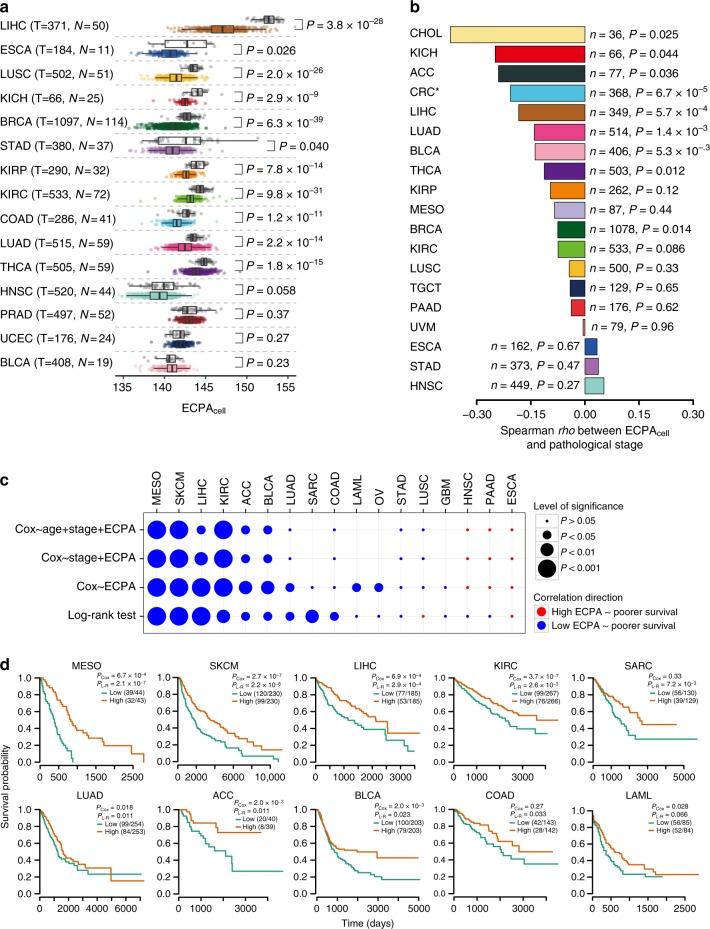
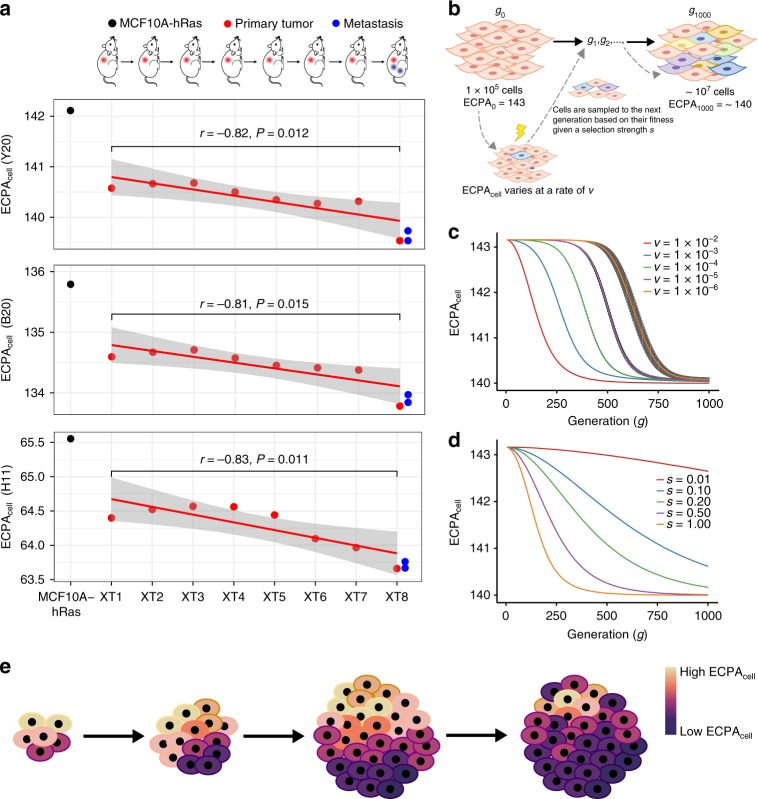
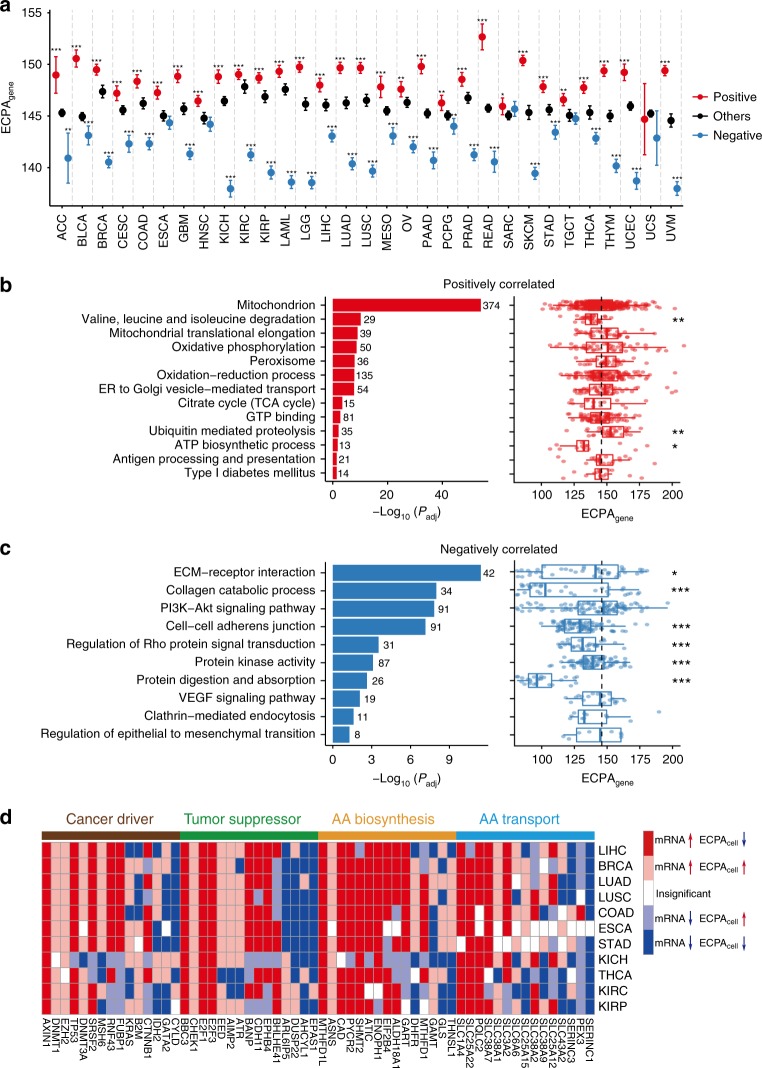
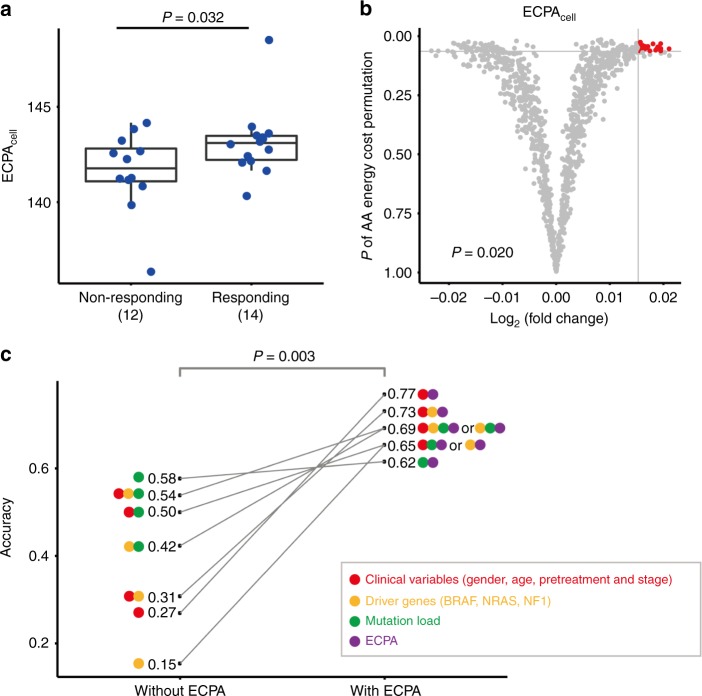
Rapidly proliferating cancer cells have much higher demand for proteinogenic amino acids than normal cells. The use of amino acids in human proteomes is largely affected by their bioavailability, which is constrained by the biosynthetic energy cost in living organisms. Conceptually distinct from gene-based analyses, we introduce the energy cost per amino acid (ECPA) to quantitatively characterize the use of 20 amino acids during protein synthesis in human cells. By analyzing gene expression data from The Cancer Genome Atlas, we find that cancer cells evolve to utilize amino acids more economically by optimizing gene expression profile and ECPA shows robust prognostic power across many cancer types. We further validate this pattern in an experimental evolution of xenograft tumors. Our ECPA analysis reveals a common principle during cancer evolution.
Conflict of interest statement
H.L. is a shareholder and on the Scientific Advisory Board for Precision Scientific Ltd. and Eagle Nebula Inc. And all authors declare no other competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
