

SWI/SNF catalytic subunits' switch drives resistance to EZH2 inhibitors in ARID1A-mutated cells
- PMID: 30297712
- PMCID: PMC6175882
- DOI: 10.1038/s41467-018-06656-6
SWI/SNF catalytic subunits' switch drives resistance to EZH2 inhibitors in ARID1A-mutated cells
Abstract
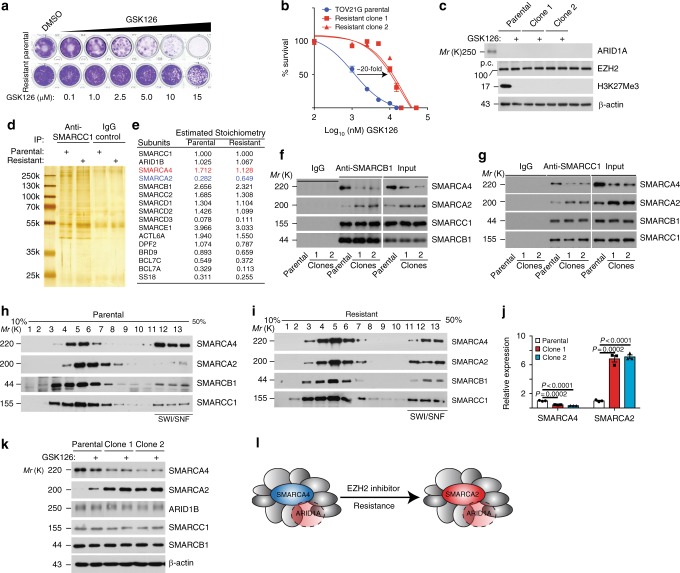
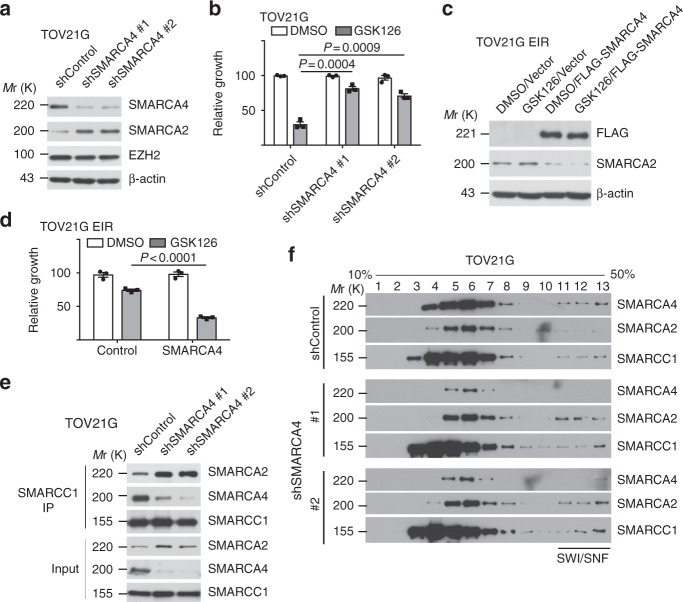
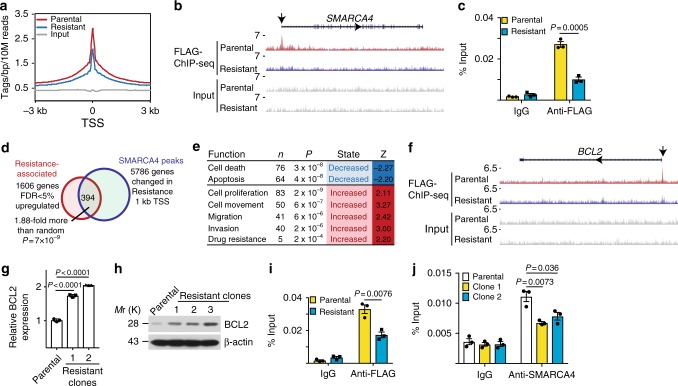
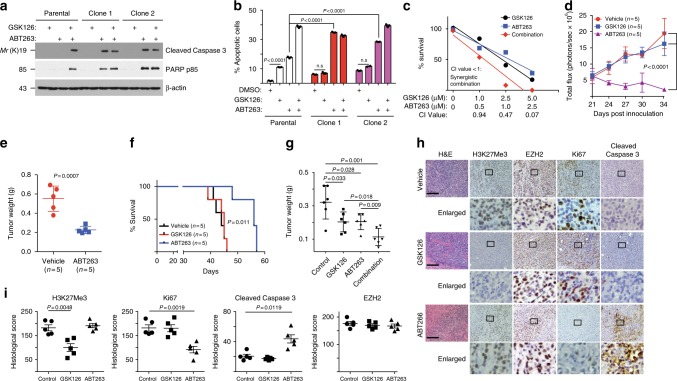
Inactivation of the subunits of SWI/SNF complex such as ARID1A is synthetically lethal with inhibition of EZH2 activity. However, mechanisms of de novo resistance to EZH2 inhibitors in cancers with inactivating SWI/SNF mutations are unknown. Here we show that the switch of the SWI/SNF catalytic subunits from SMARCA4 to SMARCA2 drives resistance to EZH2 inhibitors in ARID1A-mutated cells. SMARCA4 loss upregulates anti-apoptotic genes in the EZH2 inhibitor-resistant cells. EZH2 inhibitor-resistant ARID1A-mutated cells are hypersensitive to BCL2 inhibitors such as ABT263. ABT263 is sufficient to overcome resistance to an EZH2 inhibitor. In addition, ABT263 synergizes with an EZH2 inhibitor in vivo in ARID1A-inactivated ovarian tumor mouse models. Together, these data establish that the switch of the SWI/SNF catalytic subunits from SMARCA4 to SMARCA2 underlies the acquired resistance to EZH2 inhibitors. They suggest BCL2 inhibition alone or in combination with EZH2 inhibition represents urgently needed therapeutic strategy for ARID1A-mutated cancers.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
- S10 OD021669/OD/NIH HHS/United States
- R00 CA194318/CA/NCI NIH HHS/United States
- P30 CA010815/CA/NCI NIH HHS/United States
- R01CA160331/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)/International
- R50 CA221838/CA/NCI NIH HHS/United States
- R50 CA211199/CA/NCI NIH HHS/United States
- R01 CA202919/CA/NCI NIH HHS/United States
- R01 CA160331/CA/NCI NIH HHS/United States
- R01 CA131582/CA/NCI NIH HHS/United States
- R01 CA163377/CA/NCI NIH HHS/United States
- OC150446/DOD | Congressionally Directed Medical Research Programs (CDMRP)/International
- R01CA202919/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)/International
- R01CA163377/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)/International
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
