

Functional architecture of low-frequency variants highlights strength of negative selection across coding and non-coding annotations
- PMID: 30297966
- PMCID: PMC6236676
- DOI: 10.1038/s41588-018-0231-8
Functional architecture of low-frequency variants highlights strength of negative selection across coding and non-coding annotations
Abstract
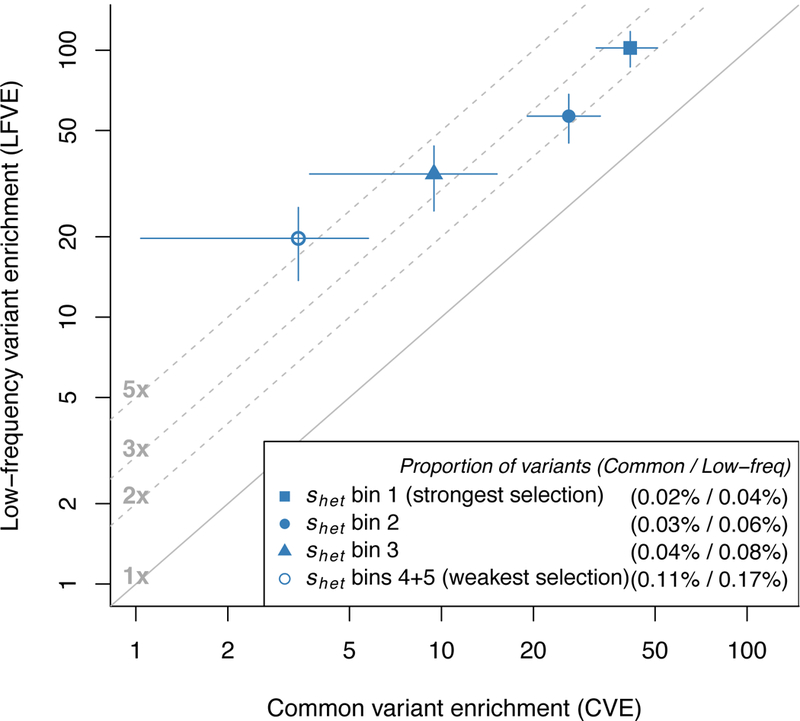
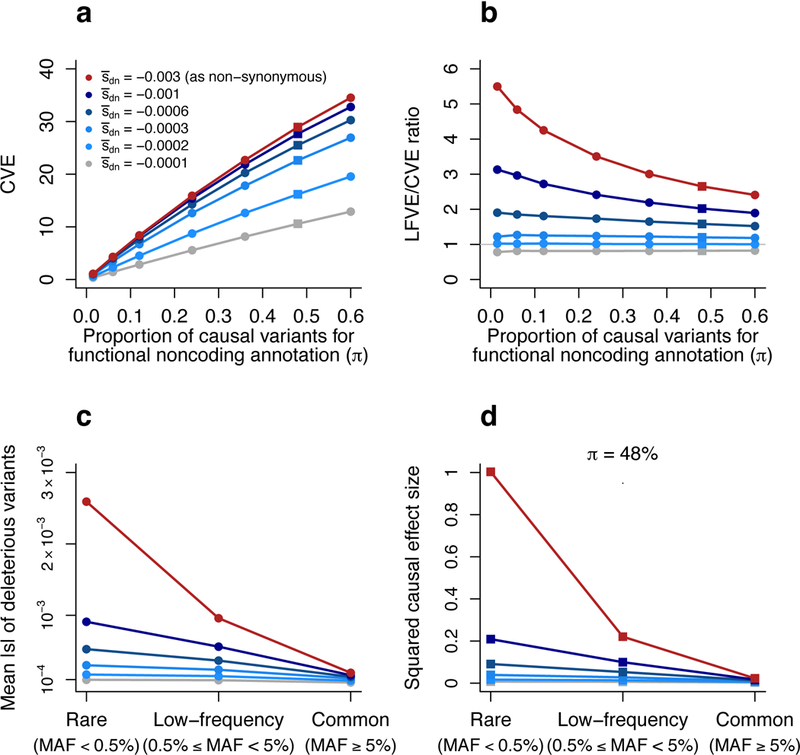
Common variant heritability has been widely reported to be concentrated in variants within cell-type-specific non-coding functional annotations, but little is known about low-frequency variant functional architectures. We partitioned the heritability of both low-frequency (0.5%≤ minor allele frequency <5%) and common (minor allele frequency ≥5%) variants in 40 UK Biobank traits across a broad set of functional annotations. We determined that non-synonymous coding variants explain 17 ± 1% of low-frequency variant heritability ([Formula: see text]) versus 2.1 ± 0.2% of common variant heritability ([Formula: see text]). Cell-type-specific non-coding annotations that were significantly enriched for [Formula: see text] of corresponding traits were similarly enriched for [Formula: see text] for most traits, but more enriched for brain-related annotations and traits. For example, H3K4me3 marks in brain dorsolateral prefrontal cortex explain 57 ± 12% of [Formula: see text] versus 12 ± 2% of [Formula: see text] for neuroticism. Forward simulations confirmed that low-frequency variant enrichment depends on the mean selection coefficient of causal variants in the annotation, and can be used to predict effect size variance of causal rare variants (minor allele frequency <0.5%).
Conflict of interest statement
Competing Financial Interests Statement
The authors declare no conflict of interest.
Figures
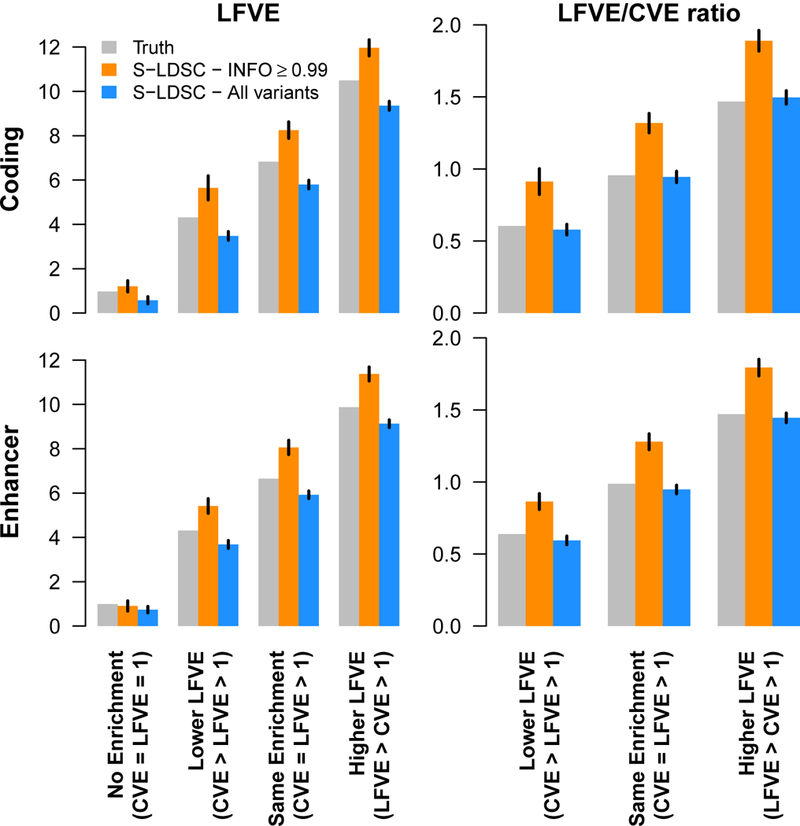
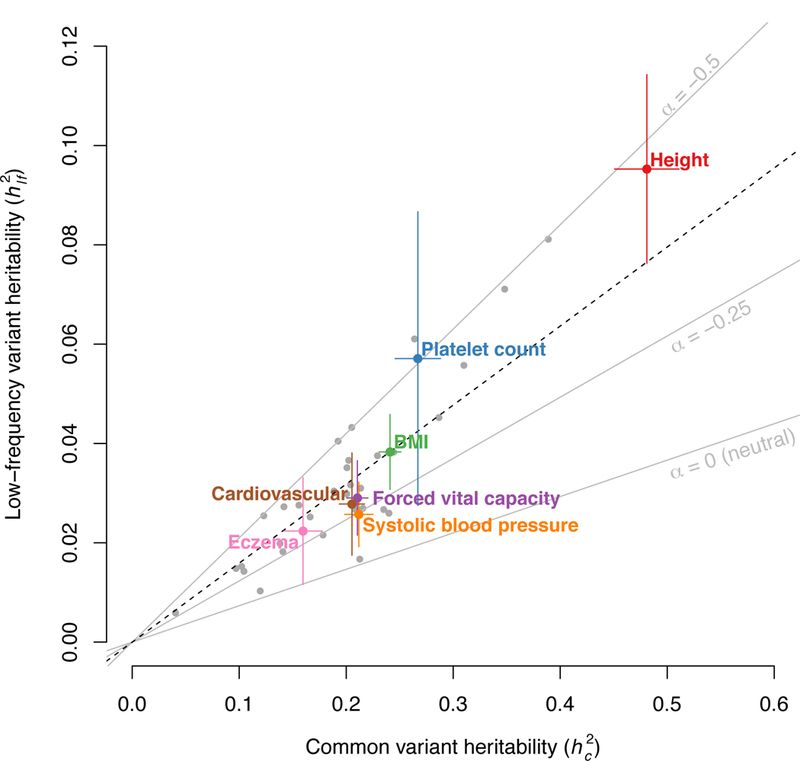
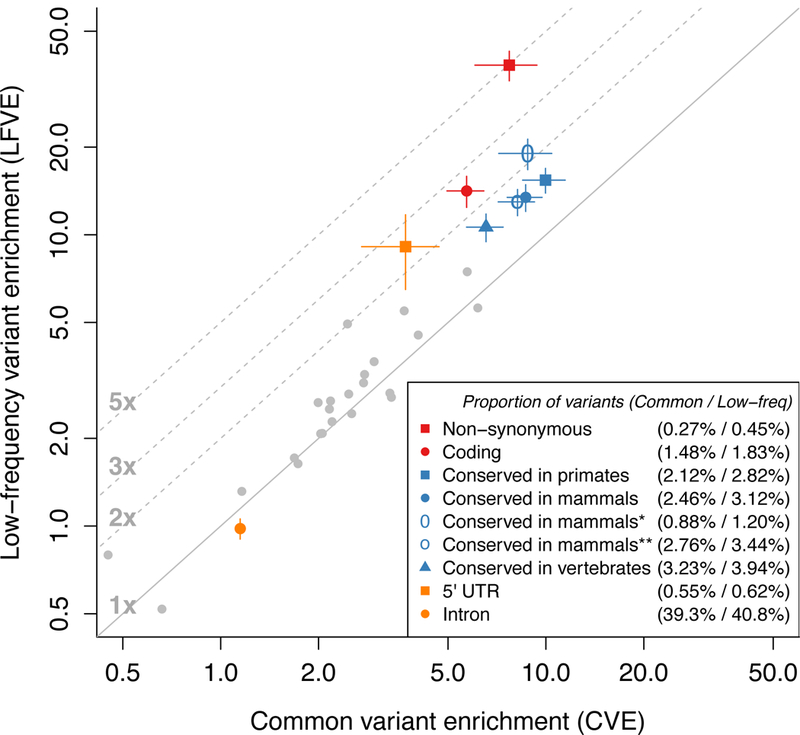
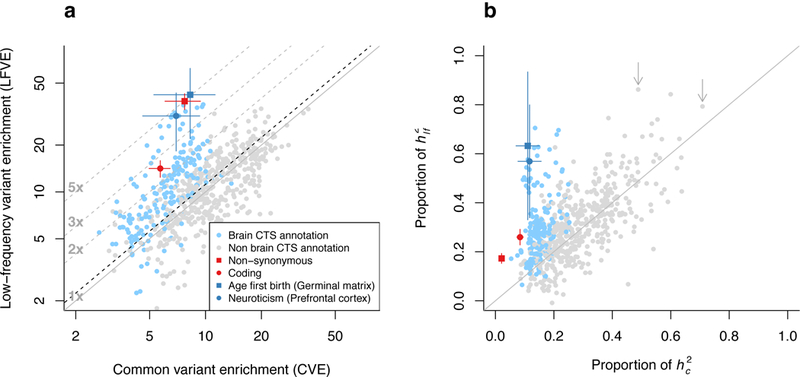
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
