

Integrated Analysis of Genetic Ancestry and Genomic Alterations across Cancers
- PMID: 30300578
- PMCID: PMC6348897
- DOI: 10.1016/j.ccell.2018.08.019
Integrated Analysis of Genetic Ancestry and Genomic Alterations across Cancers
Abstract
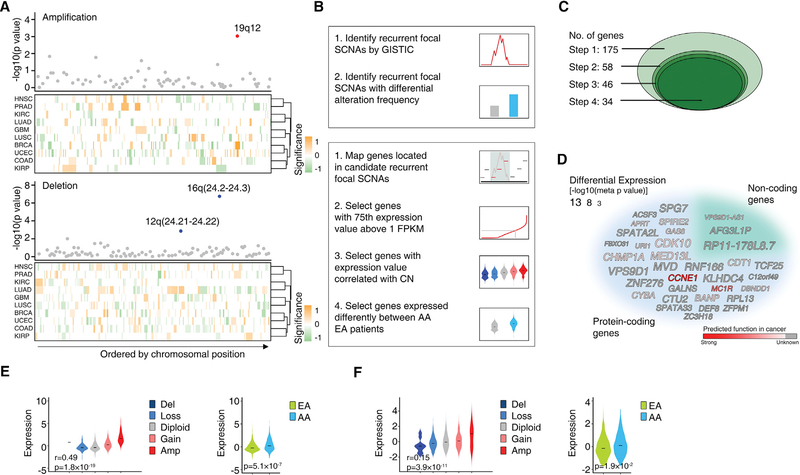
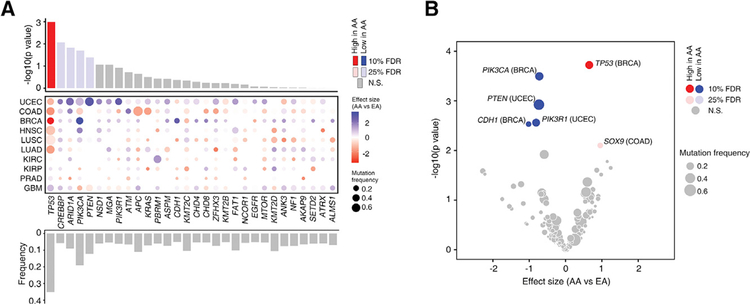
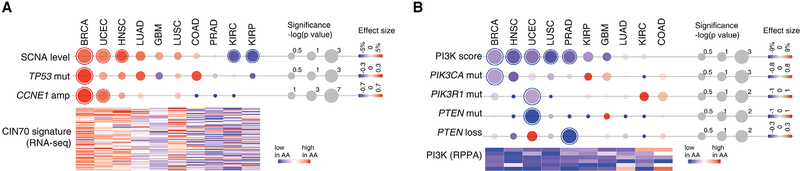
Disparities in cancer care have been a long-standing challenge. We estimated the genetic ancestry of The Cancer Genome Atlas patients, and performed a pan-cancer analysis on the influence of genetic ancestry on genomic alterations. Compared with European Americans, African Americans (AA) with breast, head and neck, and endometrial cancers exhibit a higher level of chromosomal instability, while a lower level of chromosomal instability was observed in AAs with kidney cancers. The frequencies of TP53 mutations and amplification of CCNE1 were increased in AAs in the cancer types showing higher levels of chromosomal instability. We observed lower frequencies of genomic alterations affecting genes in the PI3K pathway in AA patients across cancers. Our result provides insight into genomic contribution to cancer disparities.
Keywords: cancer disparities; cancer genetics; cancer genomics.
Copyright © 2018 Elsevier Inc. All rights reserved.
Conflict of interest statement
DECLARATION OF INTERESTS
The authors declare no competing interests.
Figures
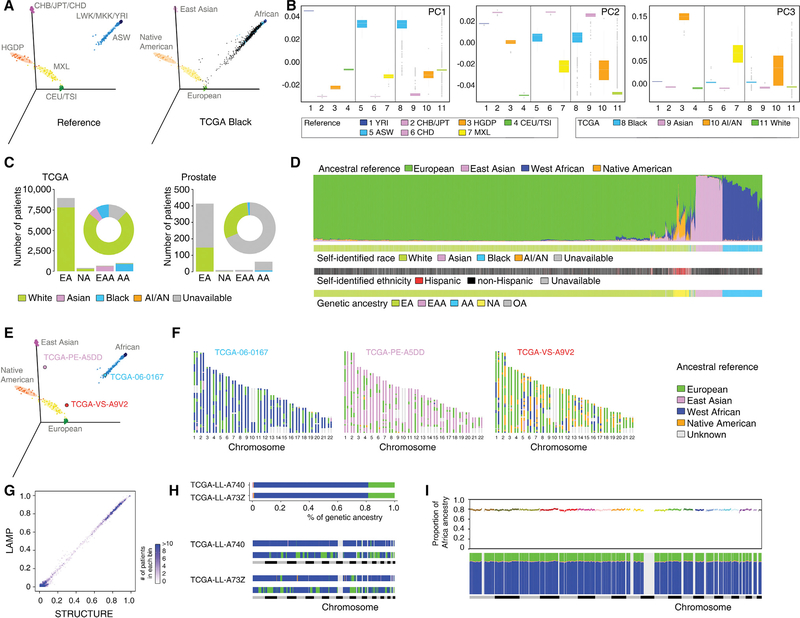
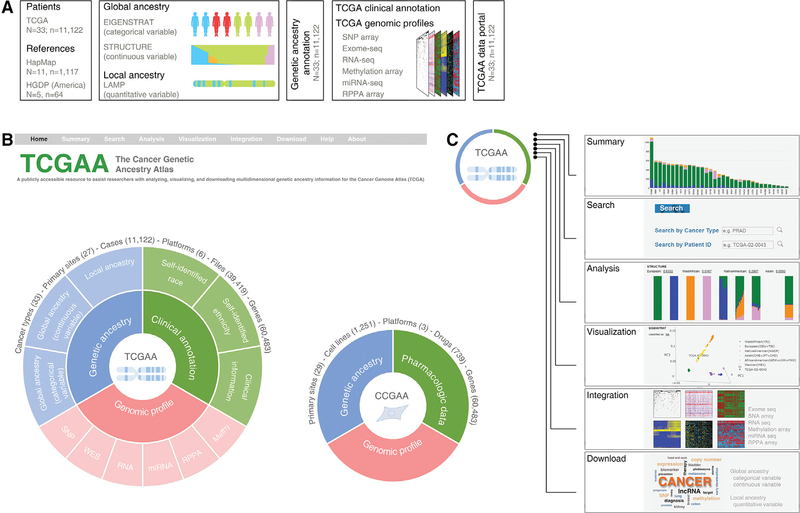
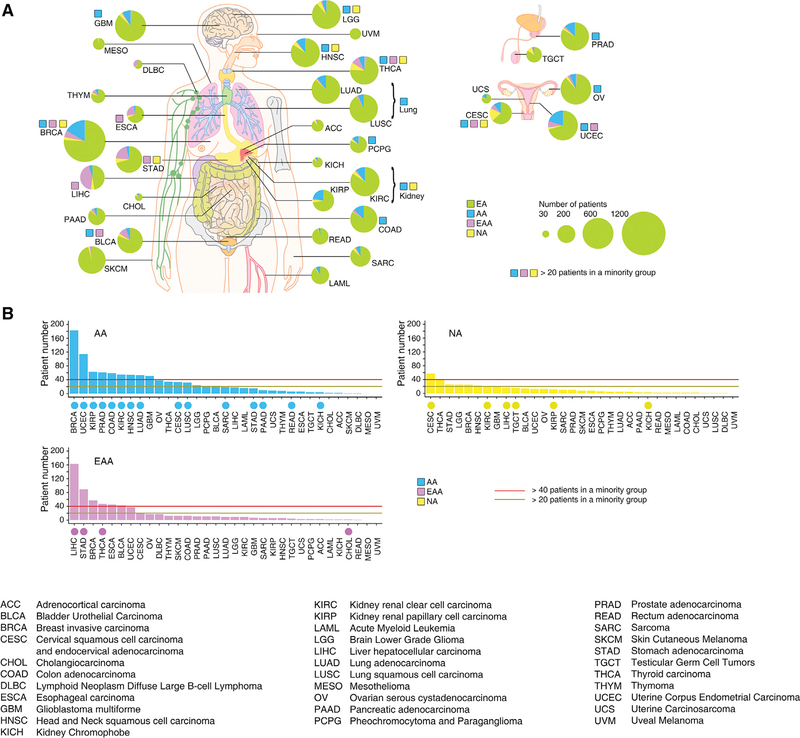
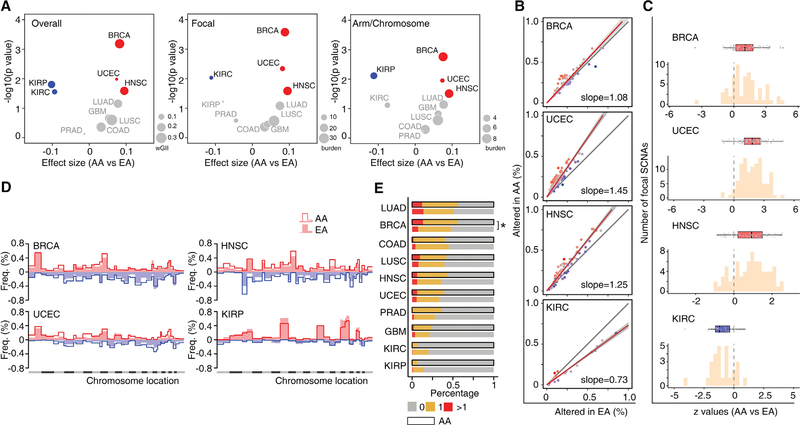
Comment in
-
Genetic Ancestry May Influence the Evolutionary Trajectory of Cancers.Cancer Cell. 2018 Oct 8;34(4):529-530. doi: 10.1016/j.ccell.2018.09.006. Cancer Cell. 2018. PMID: 30300575
References
-
- Angeloni SV, Martin MB, Garcia-Morales P, Castro-Galache MD, Ferragut JA, and Saceda M (2004). Regulation of estrogen receptor-alpha expression by the tumor suppressor gene p53 in MCF-7 cells. J. Endocrinol 180, 497–504. - PubMed
Publication types
MeSH terms
Grants and funding
- P50 CA217685/CA/NCI NIH HHS/United States
- R01 CA057341/CA/NCI NIH HHS/United States
- P30 CA010815/CA/NCI NIH HHS/United States
- R01 CA142776/CA/NCI NIH HHS/United States
- P50 CA098258/CA/NCI NIH HHS/United States
- P50 CA174523/CA/NCI NIH HHS/United States
- P50 CA083639/CA/NCI NIH HHS/United States
- U01 CA184374/CA/NCI NIH HHS/United States
- R01 CA051497/CA/NCI NIH HHS/United States
- R01 CA202919/CA/NCI NIH HHS/United States
- R01 CA190415/CA/NCI NIH HHS/United States
- T32 CA009001/CA/NCI NIH HHS/United States
- R01 CA163377/CA/NCI NIH HHS/United States
- P50 CA083638/CA/NCI NIH HHS/United States
- R01 NS094533/NS/NINDS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
