

The Oncogenic Transcription Factor RUNX1/ETO Corrupts Cell Cycle Regulation to Drive Leukemic Transformation
- PMID: 30300583
- PMCID: PMC6179967
- DOI: 10.1016/j.ccell.2018.08.015
The Oncogenic Transcription Factor RUNX1/ETO Corrupts Cell Cycle Regulation to Drive Leukemic Transformation
Erratum in
-
The Oncogenic Transcription Factor RUNX1/ETO Corrupts Cell Cycle Regulation to Drive Leukemic Transformation.Cancer Cell. 2019 Apr 15;35(4):705. doi: 10.1016/j.ccell.2019.03.012. Cancer Cell. 2019. PMID: 30991028 Free PMC article. No abstract available.
Abstract
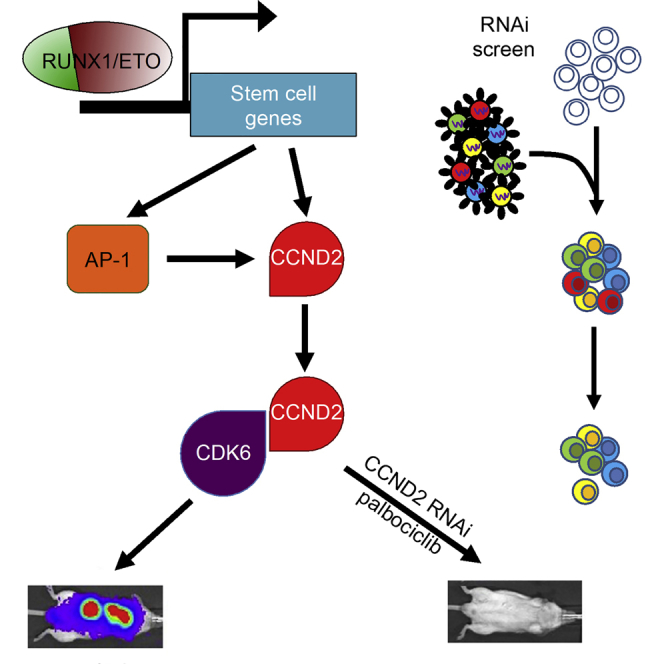
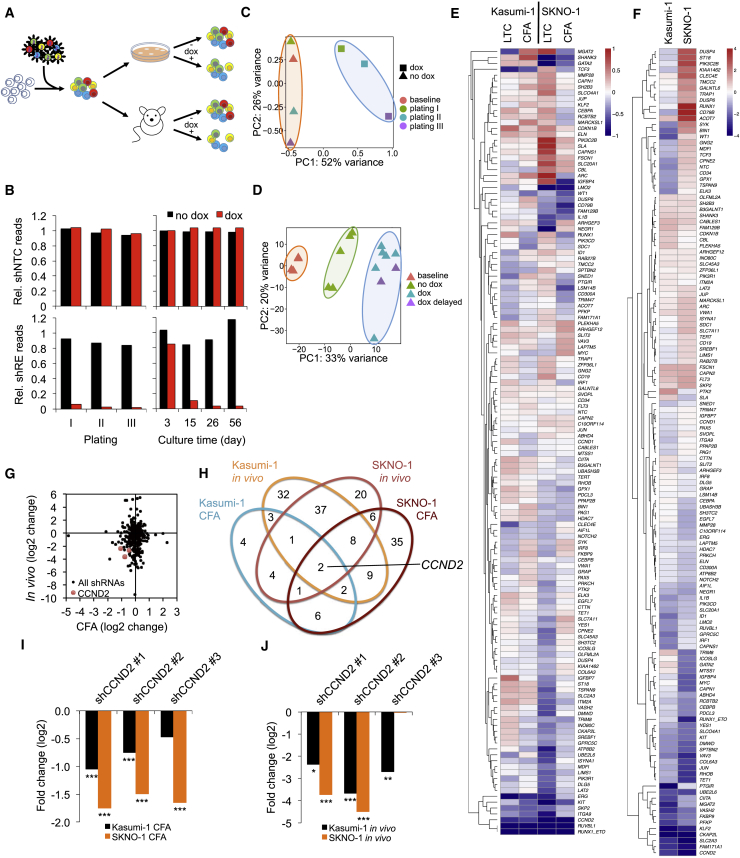
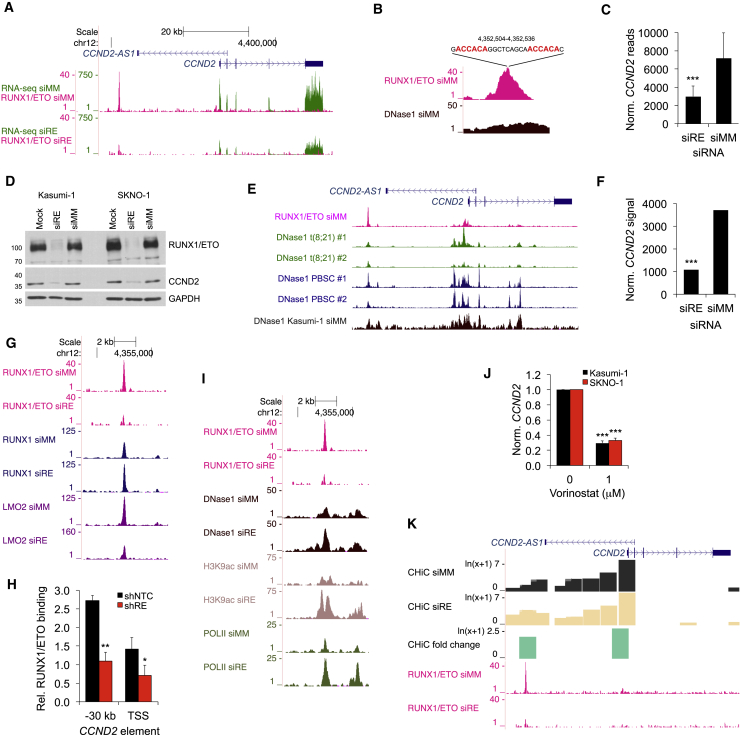
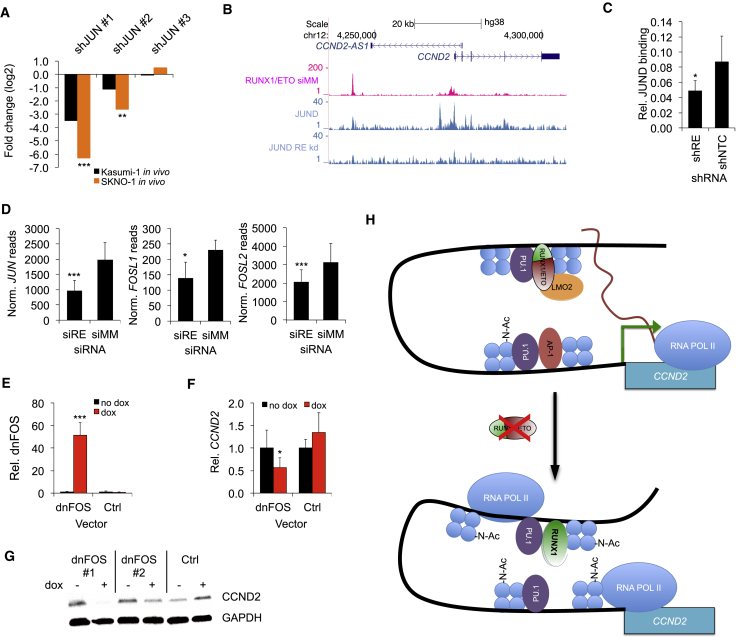
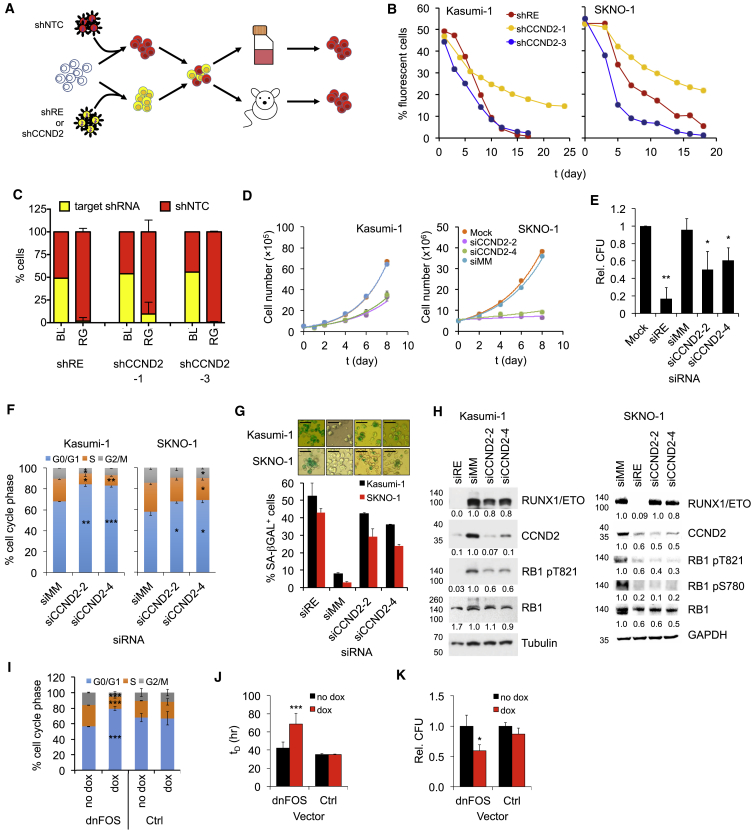
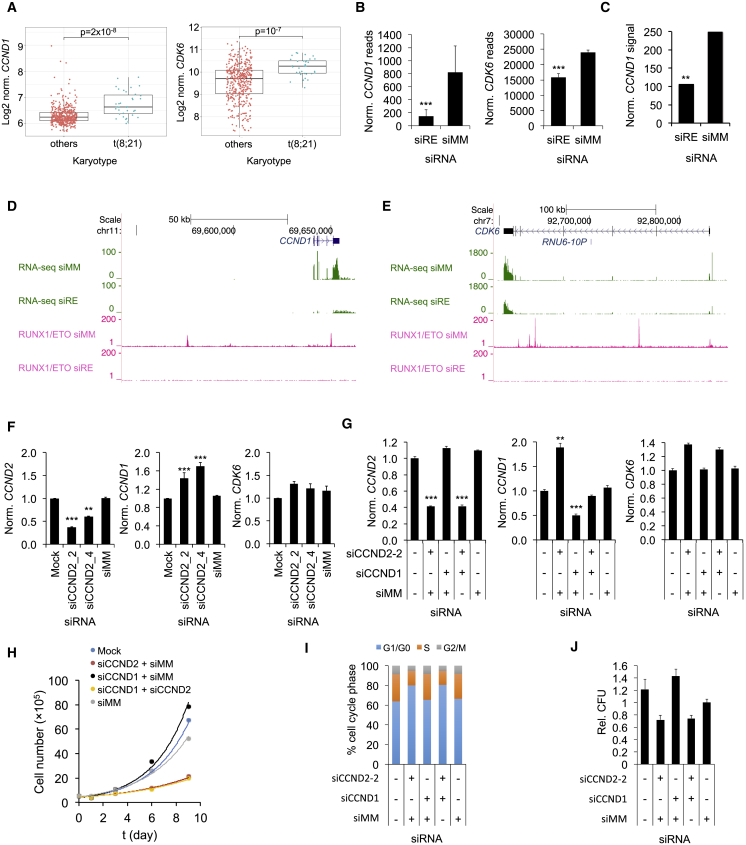
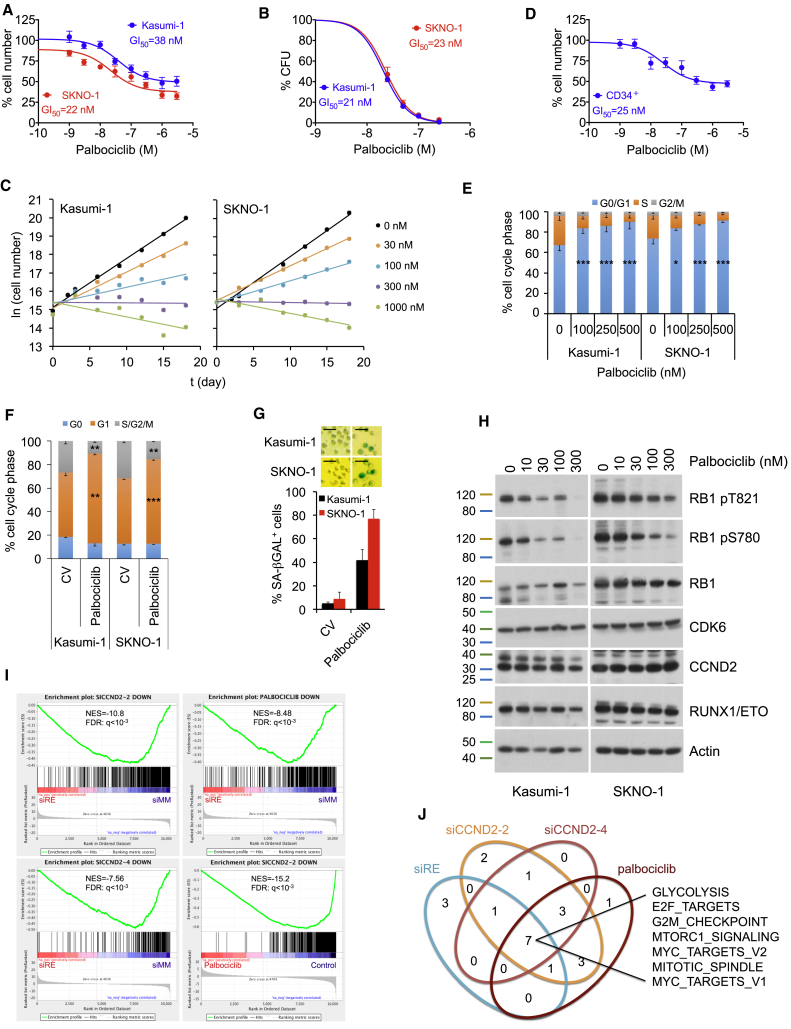
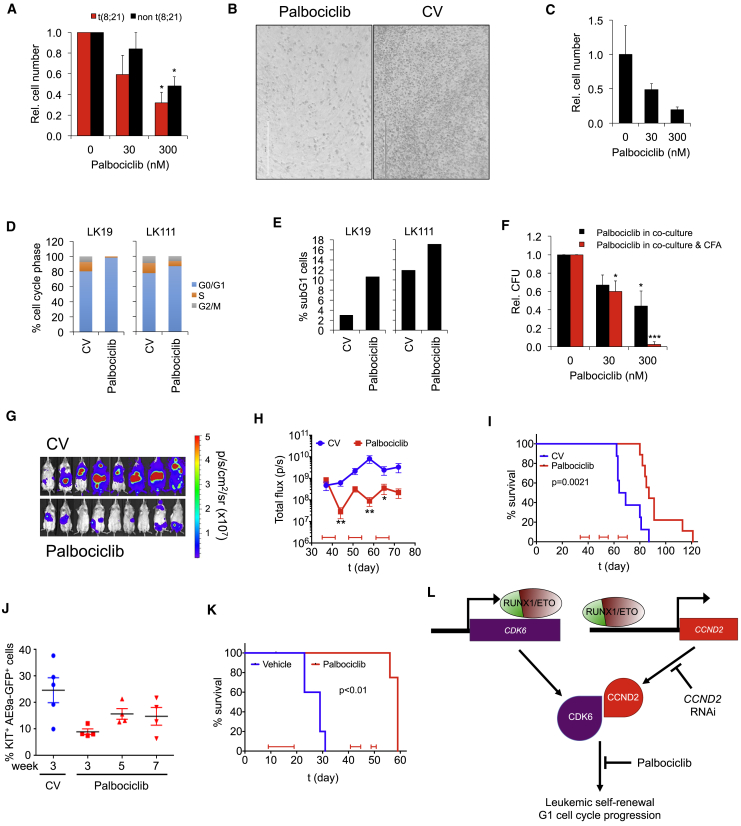
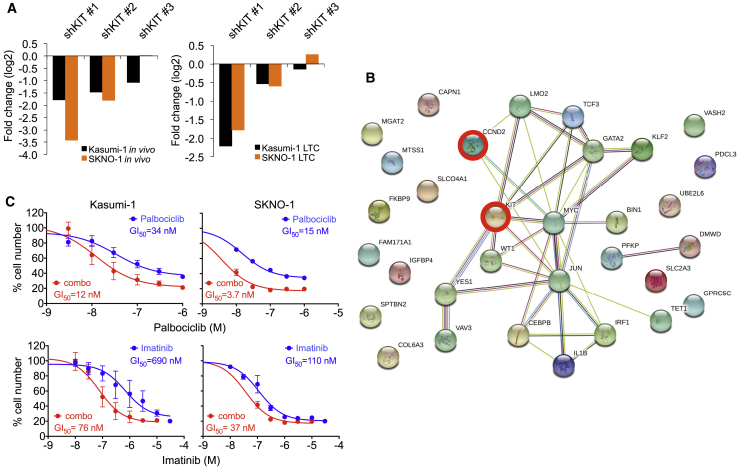
Oncogenic transcription factors such as the leukemic fusion protein RUNX1/ETO, which drives t(8;21) acute myeloid leukemia (AML), constitute cancer-specific but highly challenging therapeutic targets. We used epigenomic profiling data for an RNAi screen to interrogate the transcriptional network maintaining t(8;21) AML. This strategy identified Cyclin D2 (CCND2) as a crucial transmitter of RUNX1/ETO-driven leukemic propagation. RUNX1/ETO cooperates with AP-1 to drive CCND2 expression. Knockdown or pharmacological inhibition of CCND2 by an approved drug significantly impairs leukemic expansion of patient-derived AML cells and engraftment in immunodeficient murine hosts. Our data demonstrate that RUNX1/ETO maintains leukemia by promoting cell cycle progression and identifies G1 CCND-CDK complexes as promising therapeutic targets for treatment of RUNX1/ETO-driven AML.
Keywords: CCND2; CDK6 inhibition; KIT mutation; RNAi screen; RUNX1/ETO; acute myeloid leukemia; cell-cycle control; fusion gene; imatinib; palbociclib.
Copyright © 2018 The Author(s). Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Banker D.E., Radich J., Becker A., Kerkof K., Norwood T., Willman C., Appelbaum F.R. The t(8;21) translocation is not consistently associated with high Bcl-2 expression in de novo acute myeloid leukemias of adults. Clin. Cancer Res. 1998;4:3051–3062. - PubMed
-
- Becker H., Pfeifer D., Afonso J.D., Nimer S.D., Veelken H., Schwabe M., Lubbert M. Two cell lines of t(8;21) acute myeloid leukemia with activating KIT exon 17 mutation: models for the ‘second hit’ hypothesis. Leukemia. 2008;22:1792–1794. - PubMed
-
- Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Statist. Soc. B. 1995;57:289–300.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
