

Towards a therapy for mitochondrial disease: an update
- PMID: 30301846
- PMCID: PMC6195631
- DOI: 10.1042/BST20180134
Towards a therapy for mitochondrial disease: an update
Abstract
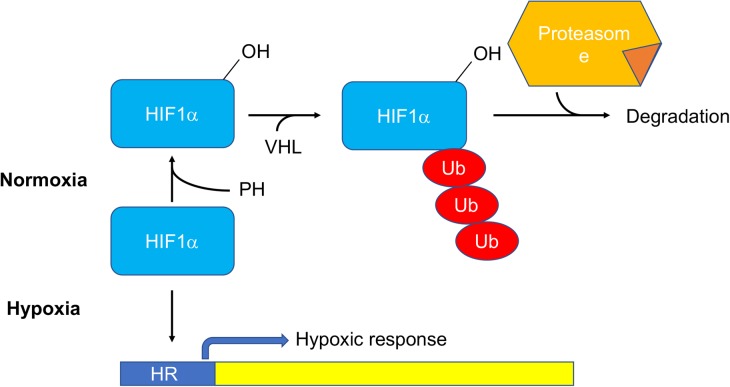
Preclinical work aimed at developing new therapies for mitochondrial diseases has recently given new hopes and opened unexpected perspectives for the patients affected by these pathologies. In contrast, only minor progresses have been achieved so far in the translation into the clinics. Many challenges are still ahead, including the need for a better characterization of the pharmacological effects of the different approaches and the design of appropriate clinical trials with robust outcome measures for this extremely heterogeneous, rare, and complex group of disorders. In this review, we will discuss the most important achievements and the major challenges in this very dynamic research field.
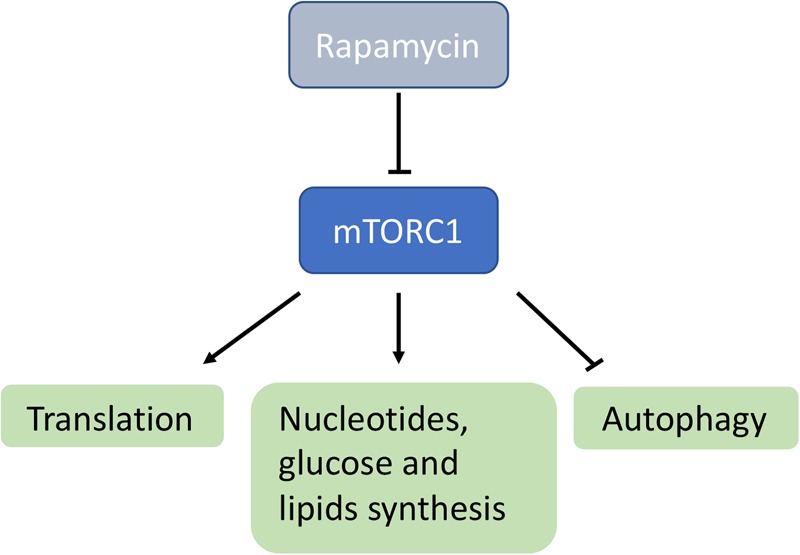
Keywords: bypass therapy; gene therapy; mitochondrial biogenesis; mitochondrial dysfunction; rapamycin.
© 2018 The Author(s).
Conflict of interest statement
C.G. shares a patented licence for the use of deoxynucleoside treatment to Meves Pharmaceuticals Inc., and may serve as a paid consultant to the same company.
Figures


References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical