

Complement Activation Contributes to Severe Acute Respiratory Syndrome Coronavirus Pathogenesis
- PMID: 30301856
- PMCID: PMC6178621
- DOI: 10.1128/mBio.01753-18
Complement Activation Contributes to Severe Acute Respiratory Syndrome Coronavirus Pathogenesis
Abstract
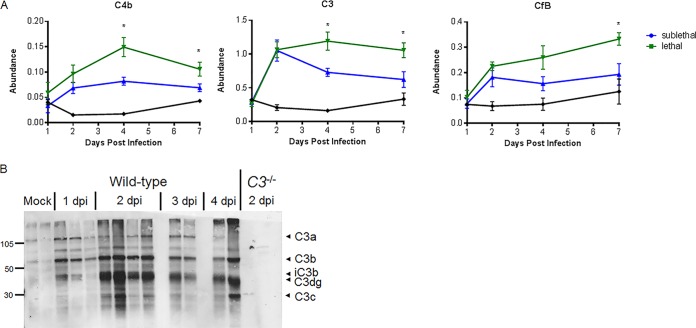
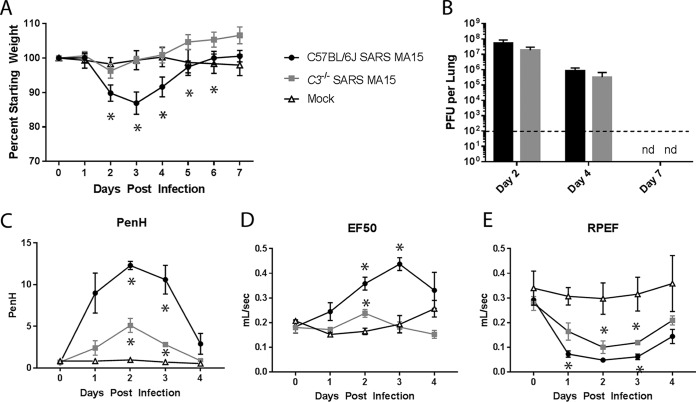
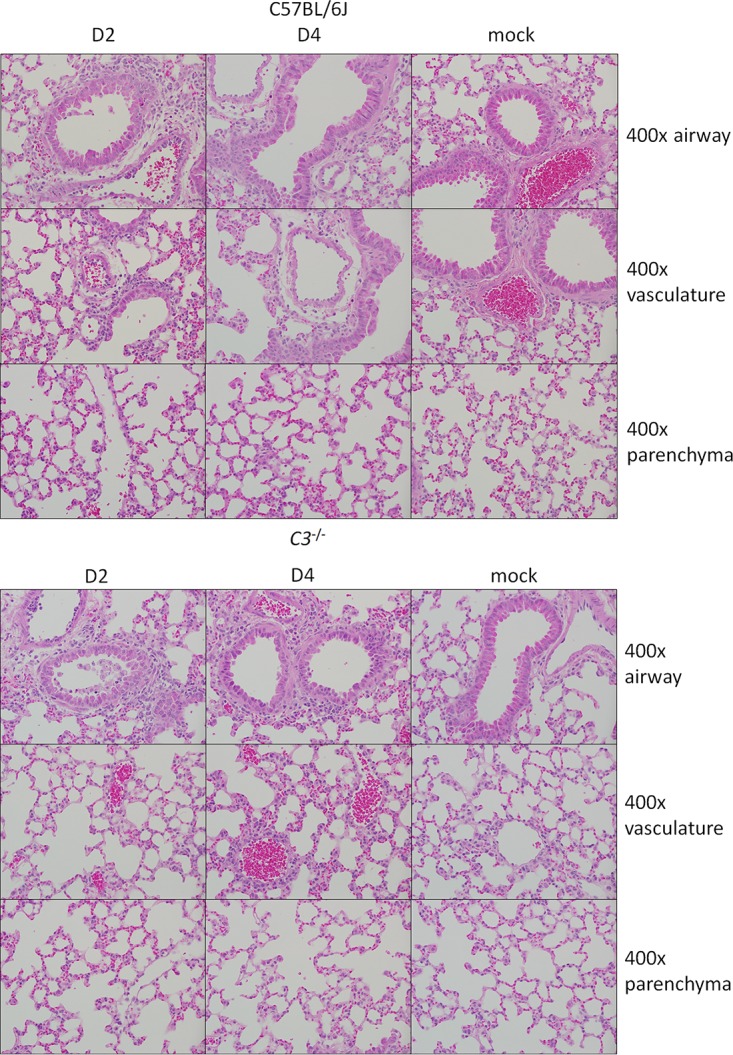
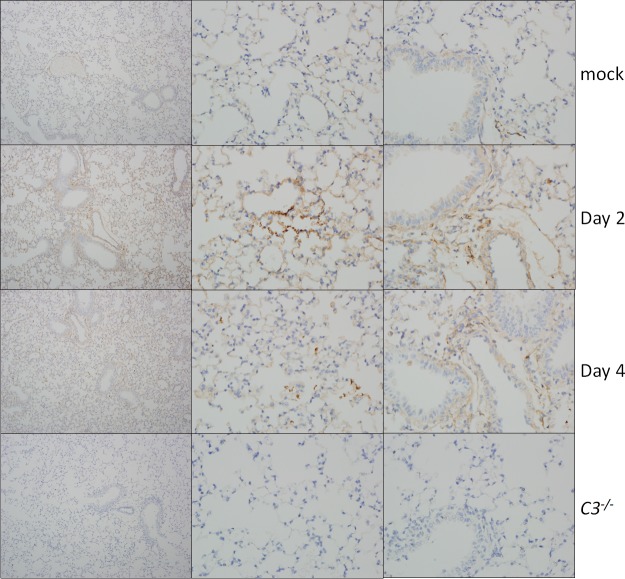
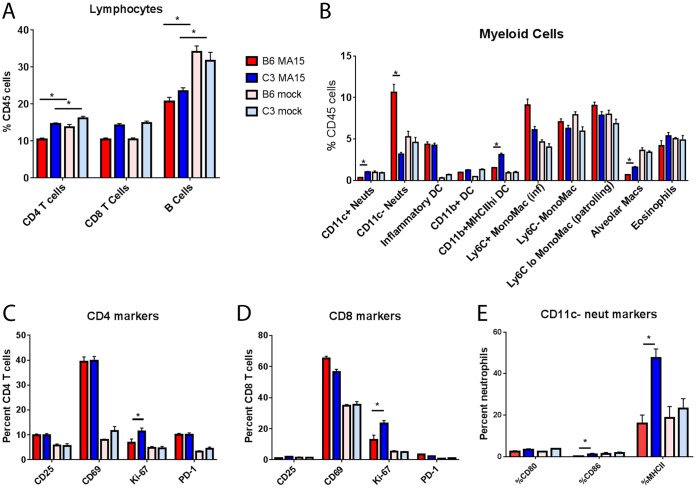
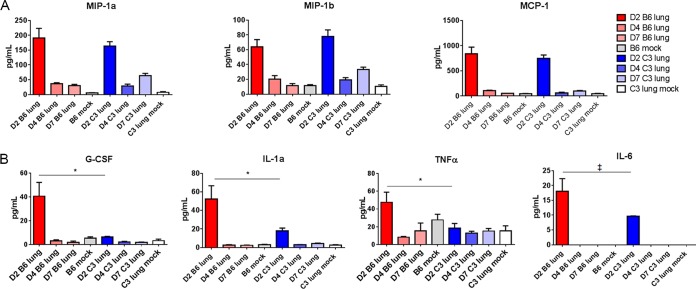
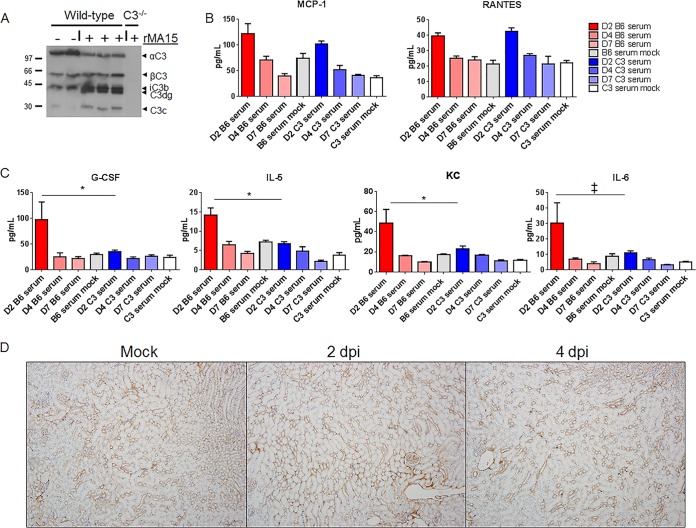
Acute respiratory distress syndrome (ARDS) is immune-driven pathologies that are observed in severe cases of severe acute respiratory syndrome coronavirus (SARS-CoV) infection. SARS-CoV emerged in 2002 to 2003 and led to a global outbreak of SARS. As with the outcome of human infection, intranasal infection of C57BL/6J mice with mouse-adapted SARS-CoV results in high-titer virus replication within the lung, induction of inflammatory cytokines and chemokines, and immune cell infiltration within the lung. Using this model, we investigated the role of the complement system during SARS-CoV infection. We observed activation of the complement cascade in the lung as early as day 1 following SARS-CoV infection. To test whether this activation contributed to protective or pathologic outcomes, we utilized mice deficient in C3 (C3-/-), the central component of the complement system. Relative to C57BL/6J control mice, SARS-CoV-infected C3-/- mice exhibited significantly less weight loss and less respiratory dysfunction despite equivalent viral loads in the lung. Significantly fewer neutrophils and inflammatory monocytes were present in the lungs of C3-/- mice than in C56BL/6J controls, and subsequent studies revealed reduced lung pathology and lower cytokine and chemokine levels in both the lungs and the sera of C3-/- mice than in controls. These studies identify the complement system as an important host mediator of SARS-CoV-induced disease and suggest that complement activation regulates a systemic proinflammatory response to SARS-CoV infection. Furthermore, these data suggest that SARS-CoV-mediated disease is largely immune driven and that inhibiting complement signaling after SARS-CoV infection might function as an effective immune therapeutic.IMPORTANCE The complement system is a critical part of host defense to many bacterial, viral, and fungal infections. It works alongside pattern recognition receptors to stimulate host defense systems in advance of activation of the adaptive immune response. In this study, we directly test the role of complement in SARS-CoV pathogenesis using a mouse model and show that respiratory disease is significantly reduced in the absence of complement even though viral load is unchanged. Complement-deficient mice have reduced neutrophilia in their lungs and reduced systemic inflammation, consistent with the observation that SARS-CoV pathogenesis is an immune-driven disease. These data suggest that inhibition of complement signaling might be an effective treatment option following coronavirus infection.
Keywords: SARS-CoV; animal models; complement; coronavirus; respiratory viruses.
Copyright © 2018 Gralinski et al.
Figures
Comment in
-
Eculizumab treatment in patients with COVID-19: preliminary results from real life ASL Napoli 2 Nord experience.Eur Rev Med Pharmacol Sci. 2020 Apr;24(7):4040-4047. doi: 10.26355/eurrev_202004_20875. Eur Rev Med Pharmacol Sci. 2020. PMID: 32329881
References
-
- Rota PA, Oberste MS, Monroe SS, Nix WA, Campagnoli R, Icenogle JP, Peñaranda S, Bankamp B, Maher K, Chen MH, Tong S, Tamin A, Lowe L, Frace M, DeRisi JL, Chen Q, Wang D, Erdman DD, Peret TC, Burns C, Ksiazek TG, Rollin PE, Sanchez A, Liffick S, Holloway B, Limor J, McCaustland K, Olsen-Rasmussen M, Fouchier R, Günther S, Osterhaus AD, Drosten C, Pallansch MA, Anderson LJ, Bellini WJ. 2003. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 300:1394–1999. doi: 10.1126/science.1085952. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
