

Uncovering association networks through an eQTL analysis involving human miRNAs and lincRNAs
- PMID: 30301969
- PMCID: PMC6177424
- DOI: 10.1038/s41598-018-33420-z
Uncovering association networks through an eQTL analysis involving human miRNAs and lincRNAs
Abstract
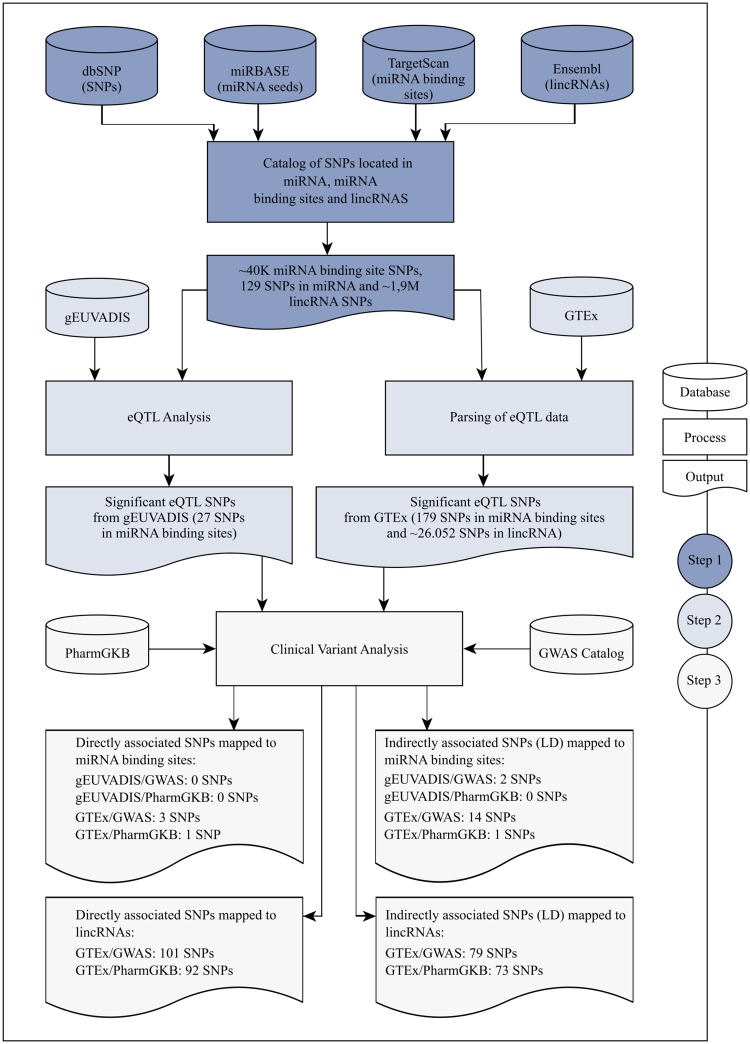
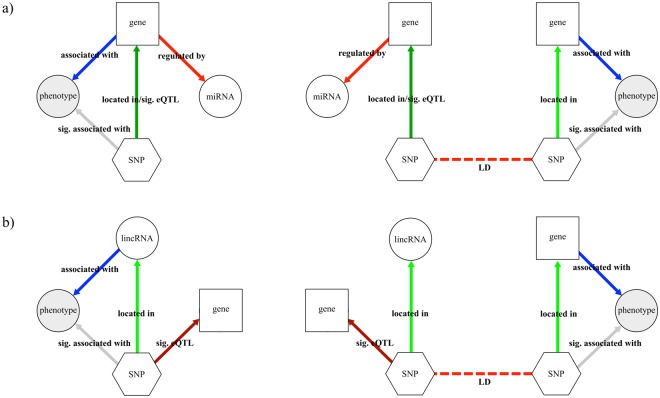
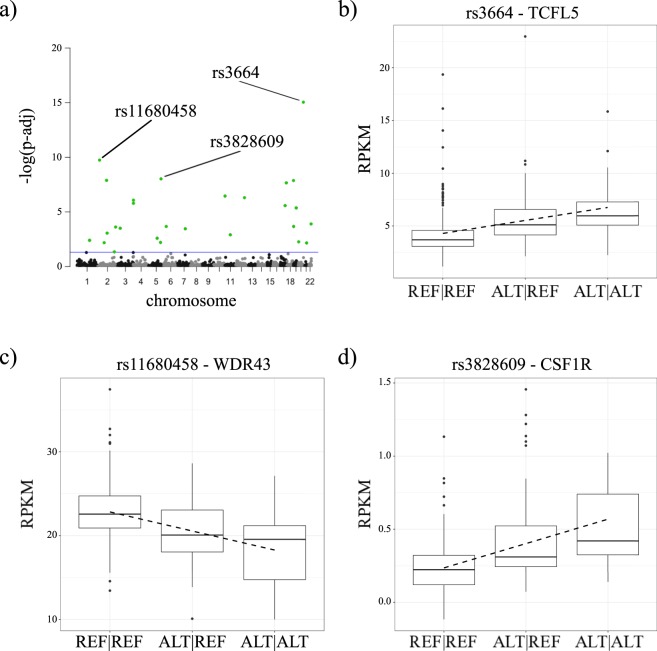
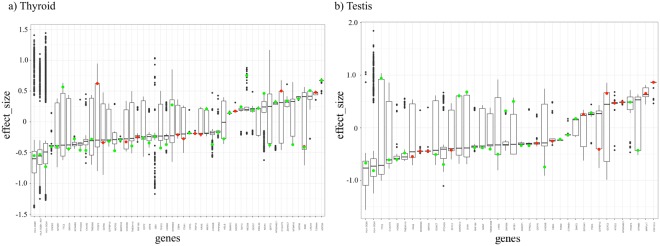
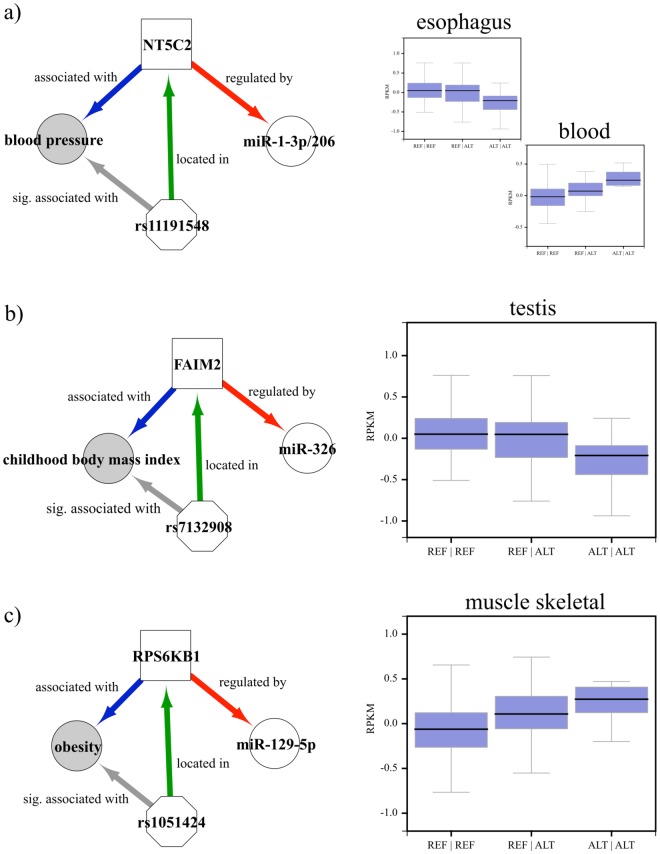
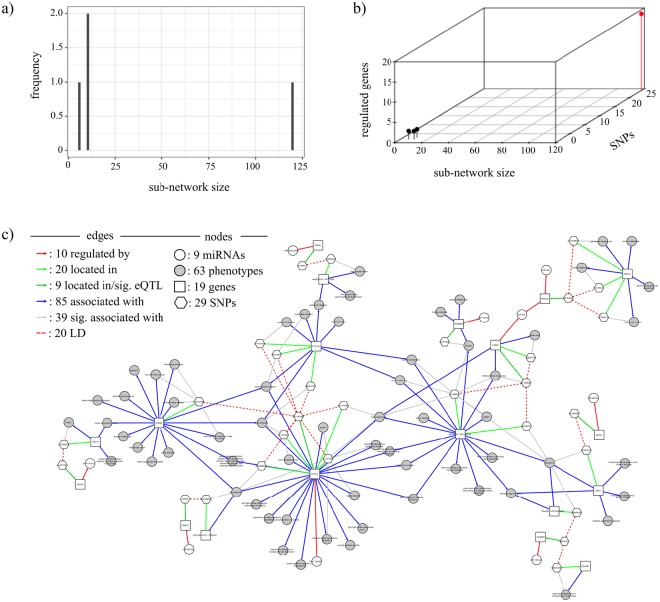
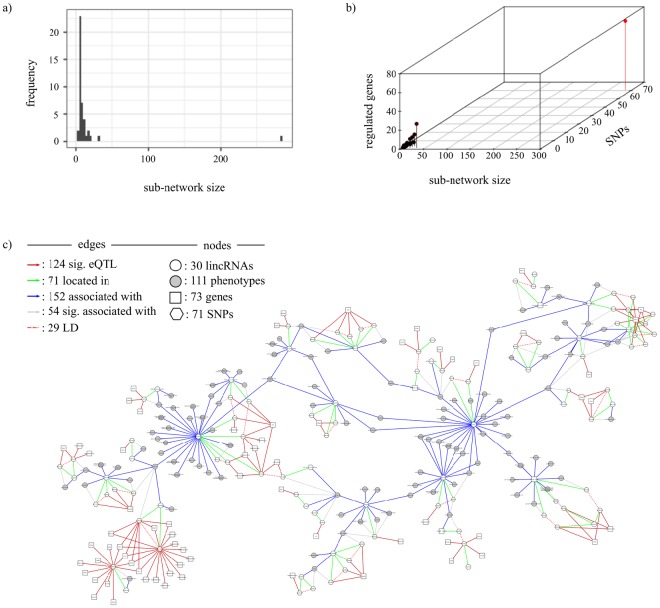
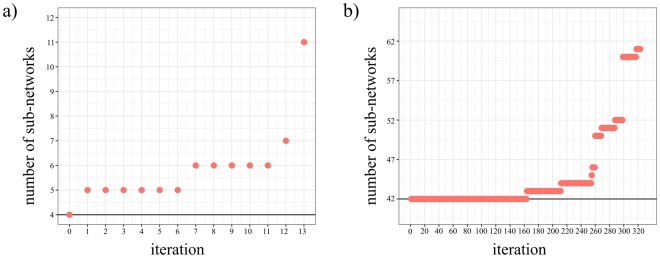
Non-coding RNAs (ncRNA) have an essential role in the complex landscape of human genetic regulatory networks. One area that is poorly explored is the effect of genetic variations on the interaction between ncRNA and their targets. By integrating a significant amount of public data, the present study cataloged the vast landscape of the regulatory effect of microRNAs (miRNA) and long intergenic noncoding RNAs (lincRNA) in the human genome. An expression quantitative trait loci (eQTL) analysis was used to identify genetic variants associated with miRNA and lincRNA and whose genotypes affect gene expression. Association networks were built for eQTL associated to traits of clinical and/or pharmacological relevance.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
