

The multifaceted role of autophagy in cancer and the microenvironment
- PMID: 30302772
- PMCID: PMC6585651
- DOI: 10.1002/med.21531
The multifaceted role of autophagy in cancer and the microenvironment
Abstract
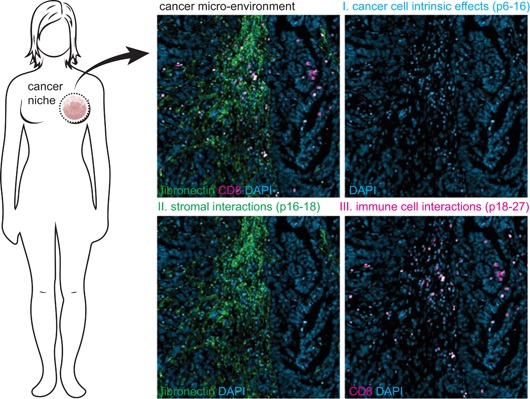
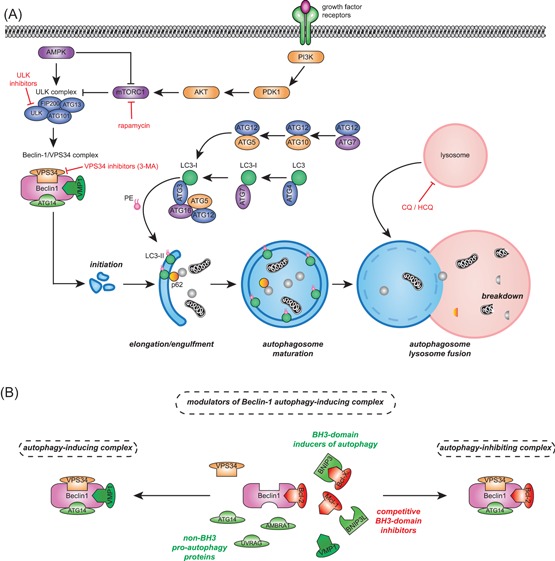
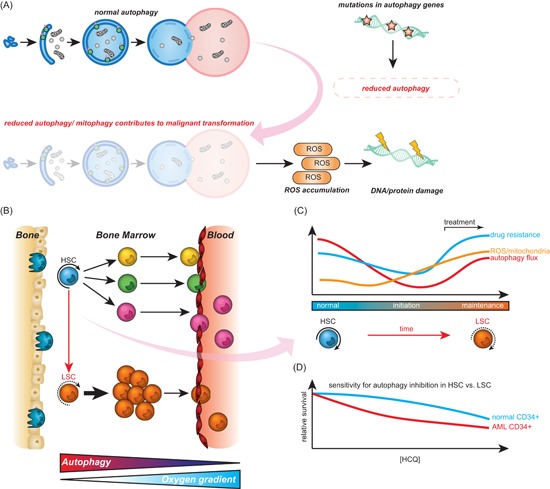
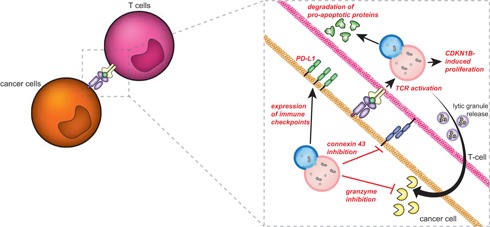
Autophagy is a crucial recycling process that is increasingly being recognized as an important factor in cancer initiation, cancer (stem) cell maintenance as well as the development of resistance to cancer therapy in both solid and hematological malignancies. Furthermore, it is being recognized that autophagy also plays a crucial and sometimes opposing role in the complex cancer microenvironment. For instance, autophagy in stromal cells such as fibroblasts contributes to tumorigenesis by generating and supplying nutrients to cancerous cells. Reversely, autophagy in immune cells appears to contribute to tumor-localized immune responses and among others regulates antigen presentation to and by immune cells. Autophagy also directly regulates T and natural killer cell activity and is required for mounting T-cell memory responses. Thus, within the tumor microenvironment autophagy has a multifaceted role that, depending on the context, may help drive tumorigenesis or may help to support anticancer immune responses. This multifaceted role should be taken into account when designing autophagy-based cancer therapeutics. In this review, we provide an overview of the diverse facets of autophagy in cancer cells and nonmalignant cells in the cancer microenvironment. Second, we will attempt to integrate and provide a unified view of how these various aspects can be therapeutically exploited for cancer therapy.
Keywords: autophagy; cancer; immune cells; microenvironment; stroma; therapy.
© 2018 The Authors. Medicinal Research Reviews Published by Wiley Periodicals, Inc.
Figures

References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
