

Recombinant GII.P16/GII.4 Sydney 2012 Was the Dominant Norovirus Identified in Australia and New Zealand in 2017
- PMID: 30304780
- PMCID: PMC6213408
- DOI: 10.3390/v10100548
Recombinant GII.P16/GII.4 Sydney 2012 Was the Dominant Norovirus Identified in Australia and New Zealand in 2017
Abstract
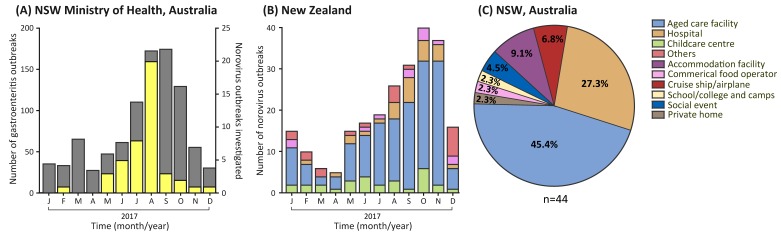
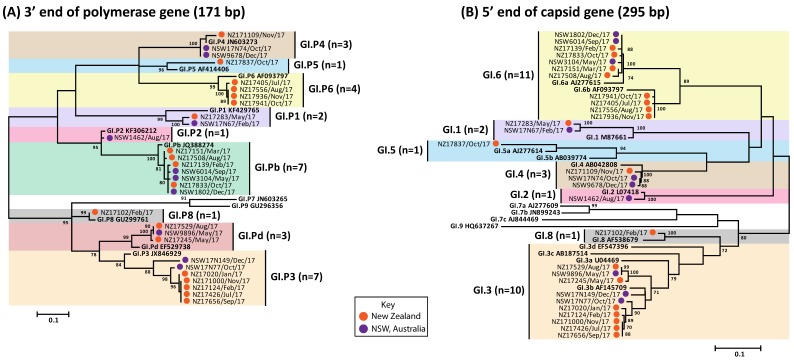
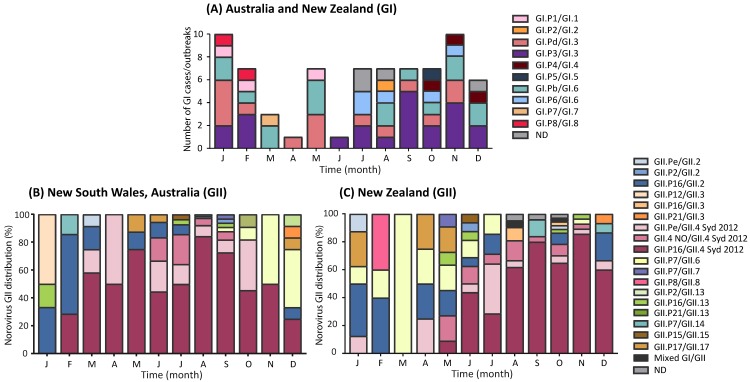
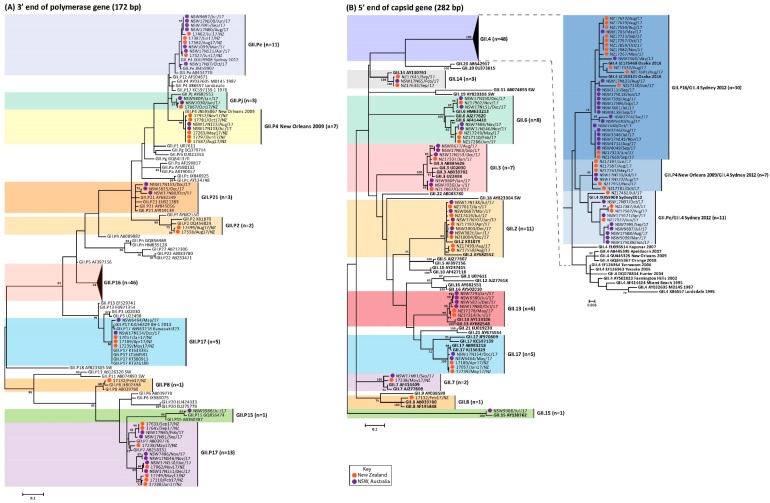
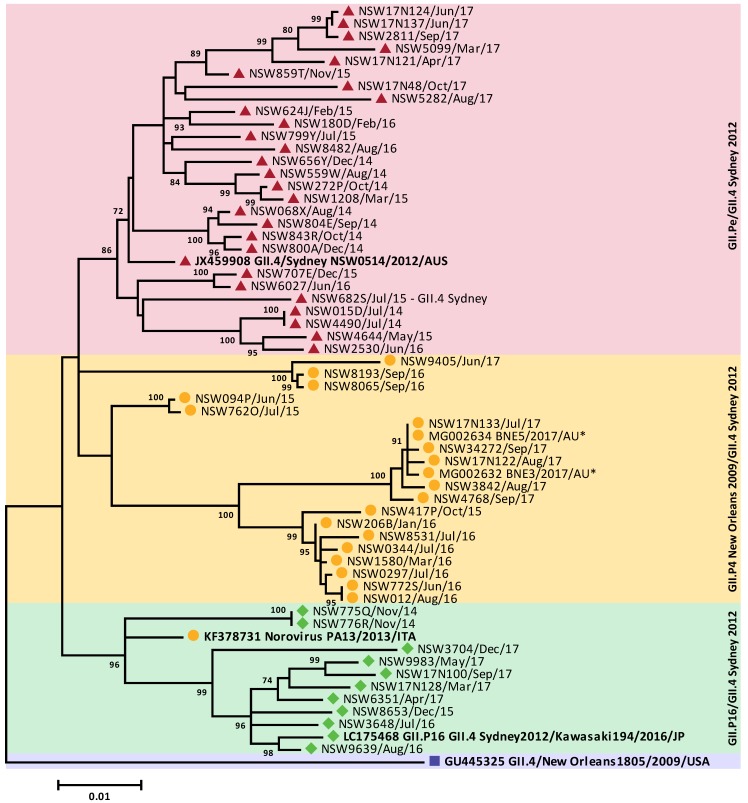
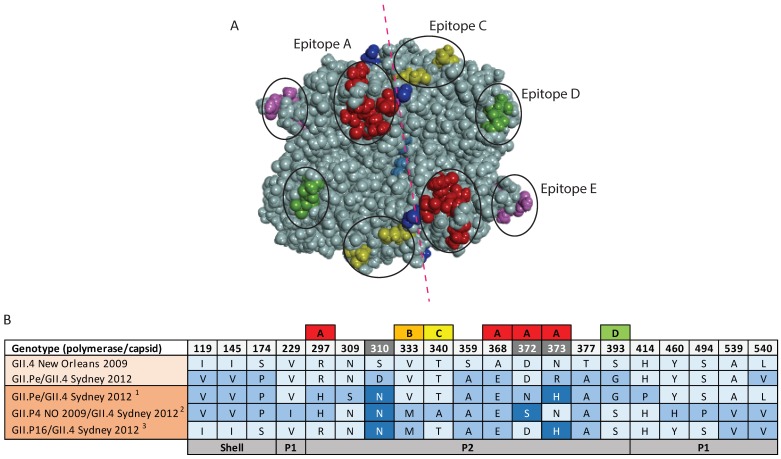
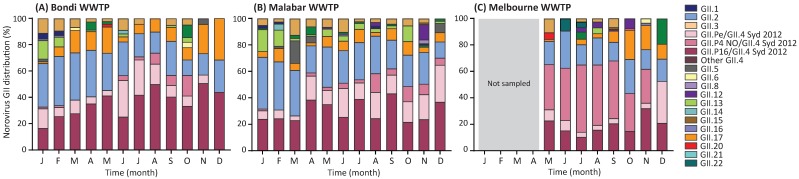
For the past two decades, norovirus pandemic variants have emerged every 3⁻5 years, and dominate until they are replaced by alternate strains. However, this scenario changed in 2016 with the co-circulation of six prevalent viruses, three of which possessed the pandemic GII.4 Sydney 2012 capsid. An increased number of institutional gastroenteritis outbreaks were reported within the Oceania region in mid-2017. This study identified emerging noroviruses circulating in Australia and New Zealand in 2017 to assess the changing dynamics of the virus infection. RT-PCR-based methods, next generation sequencing, and phylogenetic analyses were used to genotype noroviruses from both clinical and wastewater samples. Antigenic changes were observed between the capsid of pandemic Sydney 2012 variant and the two new Sydney recombinant viruses. The combination of these antigenic changes and the acquisition of a new ORF1 through recombination could both facilitate their ongoing persistence in the population. Overall, an increased prevalence of GII.P16/GII.4 Sydney 2012 viruses was observed in 2017, replacing the GII.P16/GII.2 recombinant that dominated in the region at the end of 2016. This shift in strain dominance was also observed in wastewater samples, demonstrating the reliability of wastewater as a molecular surveillance tool.
Keywords: Australia; MiSeq; New Zealand; clinical; genetic diversity; molecular epidemiology; next generation sequencing; norovirus; recombinant; wastewater.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Lozano R., Naghavi M., Foreman K., Lim S., Shibuya K., Aboyans V., Abraham J., Adair T., Aggarwal R., Ahn S.Y. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2095–2128. doi: 10.1016/S0140-6736(12)61728-0. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
