

De Novo Assembly of Two Swedish Genomes Reveals Missing Segments from the Human GRCh38 Reference and Improves Variant Calling of Population-Scale Sequencing Data
- PMID: 30304863
- PMCID: PMC6210158
- DOI: 10.3390/genes9100486
De Novo Assembly of Two Swedish Genomes Reveals Missing Segments from the Human GRCh38 Reference and Improves Variant Calling of Population-Scale Sequencing Data
Abstract
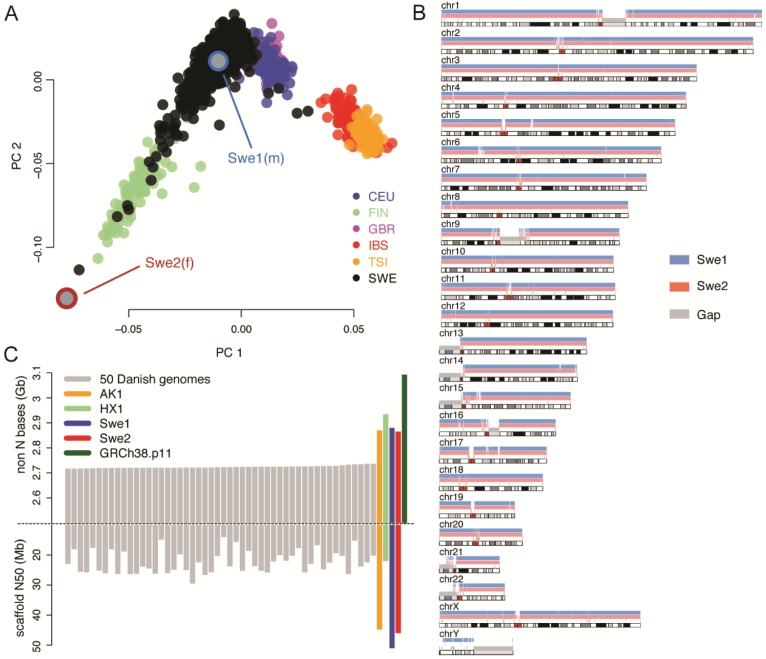
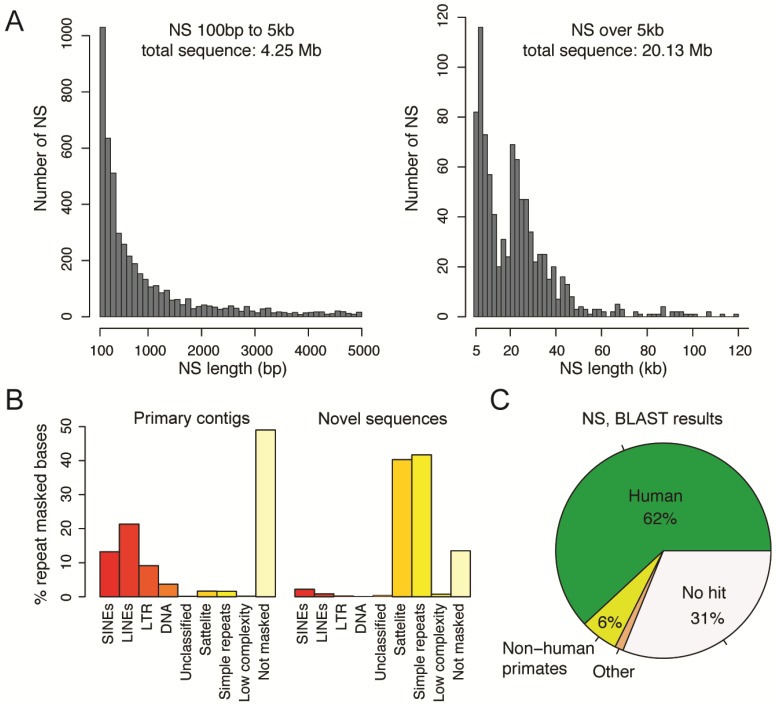
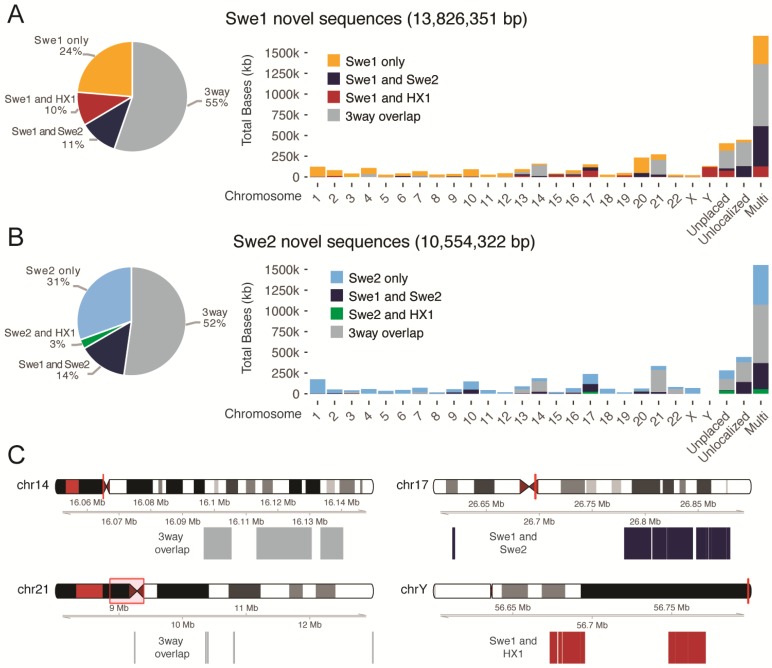
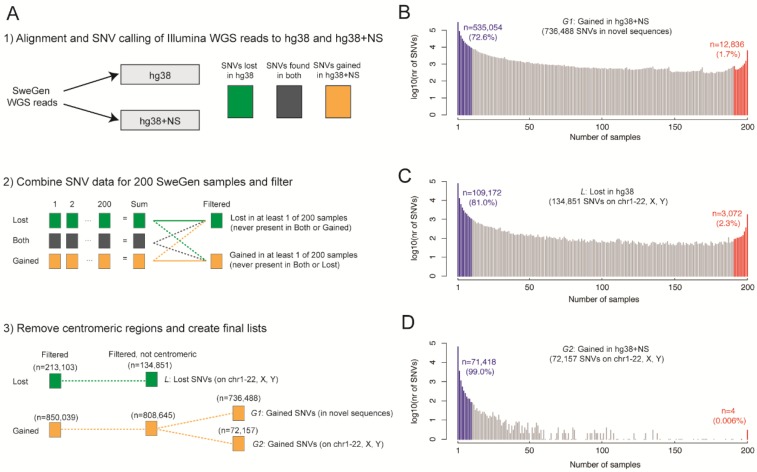
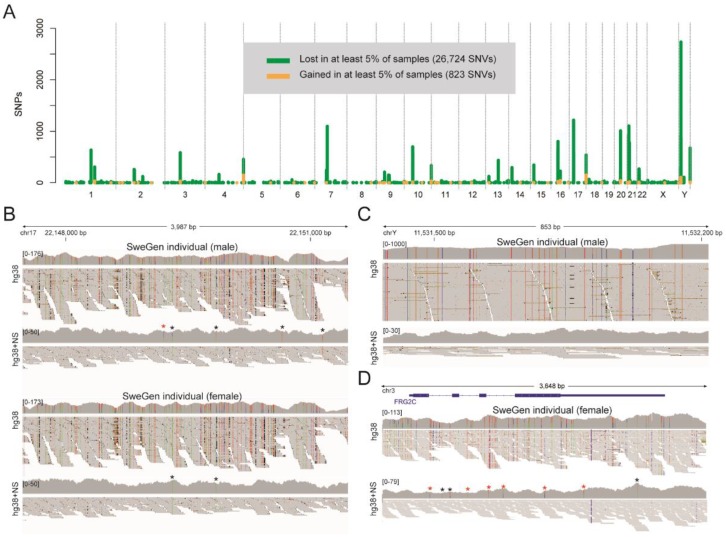
The current human reference sequence (GRCh38) is a foundation for large-scale sequencing projects. However, recent studies have suggested that GRCh38 may be incomplete and give a suboptimal representation of specific population groups. Here, we performed a de novo assembly of two Swedish genomes that revealed over 10 Mb of sequences absent from the human GRCh38 reference in each individual. Around 6 Mb of these novel sequences (NS) are shared with a Chinese personal genome. The NS are highly repetitive, have an elevated GC-content, and are primarily located in centromeric or telomeric regions. Up to 1 Mb of NS can be assigned to chromosome Y, and large segments are also missing from GRCh38 at chromosomes 14, 17, and 21. Inclusion of NS into the GRCh38 reference radically improves the alignment and variant calling from short-read whole-genome sequencing data at several genomic loci. A re-analysis of a Swedish population-scale sequencing project yields > 75,000 putative novel single nucleotide variants (SNVs) and removes > 10,000 false positive SNV calls per individual, some of which are located in protein coding regions. Our results highlight that the GRCh38 reference is not yet complete and demonstrate that personal genome assemblies from local populations can improve the analysis of short-read whole-genome sequencing data.
Keywords: GRCh38; SMRT sequencing; Swedish population; de novo assembly; human reference genome; human whole-genome sequencing; population sequencing.
Conflict of interest statement
The authors declare that they have no competing interests
Figures
References
-
- Ameur A., Dahlberg J., Olason P., Vezzi F., Karlsson R., Martin M., Viklund J., Kahari A.K., Lundin P., Che H., et al. SweGen: A whole-genome data resource of genetic variability in a cross-section of the Swedish population. Eur. J. Hum. Genet. 2017;25:1253–1260. doi: 10.1038/ejhg.2017.130. - DOI - PMC - PubMed
-
- Fakhro K.A., Staudt M.R., Ramstetter M.D., Robay A., Malek J.A., Badii R., Al-Marri A.A., Abi Khalil C., Al-Shakaki A., Chidiac O., et al. The Qatar genome: A population-specific tool for precision medicine in the Middle East. Hum. Genome Var. 2016;3:16016. doi: 10.1038/hgv.2016.16. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
