

Description of the molecular and phenotypic spectrum of Wiedemann-Steiner syndrome in Chinese patients
- PMID: 30305169
- PMCID: PMC6180513
- DOI: 10.1186/s13023-018-0909-0
Description of the molecular and phenotypic spectrum of Wiedemann-Steiner syndrome in Chinese patients
Abstract
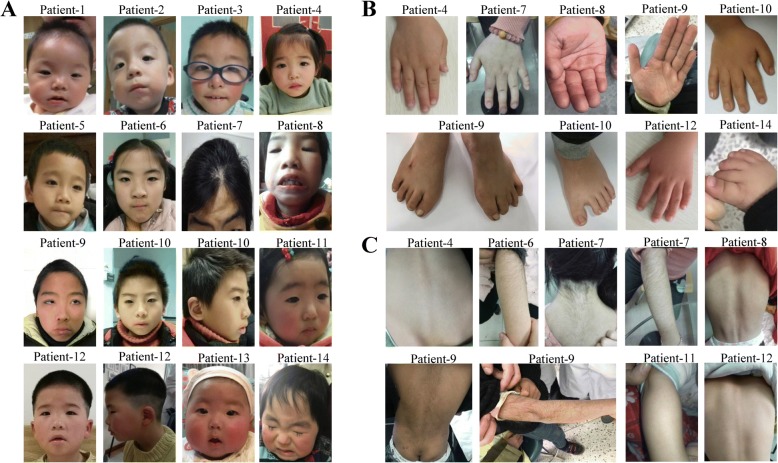
Background: Wiedemann-Steiner syndrome (WDSTS) is a rare genetic disorder characterized by facial gestalt, neurodevelopmental delay, skeletal anomalies and growth retardation, which is caused by variation of KMT2A gene. To date, only 2 Chinese WDSTS patients have been reported. Here, we report the phenotypes and KMT2A gene variations in 14 unrelated Chinese WDSTS patients and investigate the phenotypic differences between the Chinese and French cohorts.
Methods: Next generation sequencing was performed for each patient, and the variants in the KMT2A gene were validated by Sanger sequencing. The phenotypes of 16 Chinese WDSTS patients were summarized and compared to 33 French patients.
Results: Genetic sequencing identified 13 deleterious de novo KMT2A variants in 14 patients, including 10 truncating, 2 missenses and 1 splicing variants. Of the 13 variants, 11 are novel and two have been reported previously. One of the patients is mosaic in the KMT2A gene. The variation spectra and phenotypic profiles of the Chinese WDSTS patients showed no difference with patients of other ethnicities; however, differ in the frequencies of several clinical features. We demonstrated that variations in the KMT2A gene can lead to both advanced and delayed bone age. We identified 6 novel phenotypes, which include microcephaly, deep palmar crease, external ear deformity, carpal epiphyseal growth retardation, dyslipidemia, and glossoptosis. In addition, patients harbored missense variants in the CXXC zinc finger domain of KMT2A showed more severe neurophenotypes.
Conclusion: Our study consists of the largest cohort of Chinese WDSTS patients that continues to expand the WDSTS phenotypic and variation spectrum. Our results support the notion that the CXXC zinc finger domain of KMT2A gene is a hotspot for missense variants associated with more severe neurophenotypes.
Keywords: Chinese patients; KMT2A variation; Phenotypic differences; Wiedemann–Steiner syndrome.
Conflict of interest statement
Ethics approval and consent to participate
All procedures followed were in accordance with the ethical standards of the responsible institutional committee on human experimentation and with the Helsinki Declaration of 1975 (revised in 2000). The protocols were approved by the Institutional Medical Ethics Committee.
Consent for publication
Informed consent was obtained from each patient’s family.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
- 81472051/National Natural Science Foundation of China/International
- 81601869/National Natural Science Foundation of China/International
- 81772303/National Natural Science Foundation of China/International
- 15410722800/Project of Shanghai Municipal Science and Technology Commission/International
- 20152529/Project of Shanghai Municipal Education Commission- Gaofeng Clinical Medicine/International
LinkOut - more resources
Full Text Sources
