

Review
doi: 10.1038/s41576-018-0055-5.
The phylogenomics of evolving virus virulence
Affiliations
- PMID: 30305704
- PMCID: PMC7096893
- DOI: 10.1038/s41576-018-0055-5
Item in Clipboard
Review
The phylogenomics of evolving virus virulence
Nat Rev Genet.
2018 Dec.
Abstract
How virulence evolves after a virus jumps to a new host species is central to disease emergence. Our current understanding of virulence evolution is based on insights drawn from two perspectives that have developed largely independently: long-standing evolutionary theory based on limited real data examples that often lack a genomic basis, and experimental studies of virulence-determining mutations using cell culture or animal models. A more comprehensive understanding of virulence mutations and their evolution can be achieved by bridging the gap between these two research pathways through the phylogenomic analysis of virus genome sequence data as a guide to experimental study.
Conflict of interest statement
The authors declare no competing interests.
Figures

a | A model phylogeny with virulence determinants mapped to a fairly deep node suggesting that higher virulence has increased virus fitness. b | A model phylogeny with virulence traits mapped to shallow nodes suggesting that higher virulence reduced pathogen fitness so that viruses with these mutations are purged from the population or require compensatory mutations. c | A model phylogeny with a high-virulence mutation arising multiple times independently owing to parallel or convergent evolution. The occurrence of parallel/convergent mutations that occur more frequently than by chance is likely to reflect adaptive evolution (Fig. 2). d | The relationship between virulence, fitness and host jumps. A virus is assumed to be at a fitness peak (high R0), in this case high virulence, in the reservoir host, so that the mutations determining both virulence and host range are expected to be subject to strong purifying selection (for example, a low value of dN/dS). As the virus emerges in the new recipient host, it will initially be maladapted (that is, reside in a fitness valley) and subject to genetic drift as the population is small. As it adapts to the new host, virulence will be selectively optimized (in this case declining), increasing R0 and resulting in positive selection (for example, dN/dS > 1, although other measures of selection pressure are available). Once the virus becomes adapted to the new host, the virulence determinants are again subject to purifying selection.

The evolution of virulence in strains of oral polio vaccine (OPV). OPV is an attenuated form of poliovirus that can occasionally revert to a virulent form and cause outbreaks of poliomyelitis. A | Phylogenetic analysis of OPV strains in nature reveals that some mutations associated with high virulence have experienced more frequent parallel evolution than expected by chance (and occupy well supported nodes) and hence are likely to be seletively favoured. B | Computational evolutionary analysis then reveals that this parallel evolution for high virulence is associated with a hypothetical threonine-to-proline (T-to-P) amino acid change that is subject to significant adaptive evolution (which can be detected in a variety of ways),. C | The virulence impact of these mutations is then confirmed in both in vitro (cell culture; part Ca) and in vivo (mouse; part Cb) experimental studies. In all cases, the red shading signifies increased virulence.
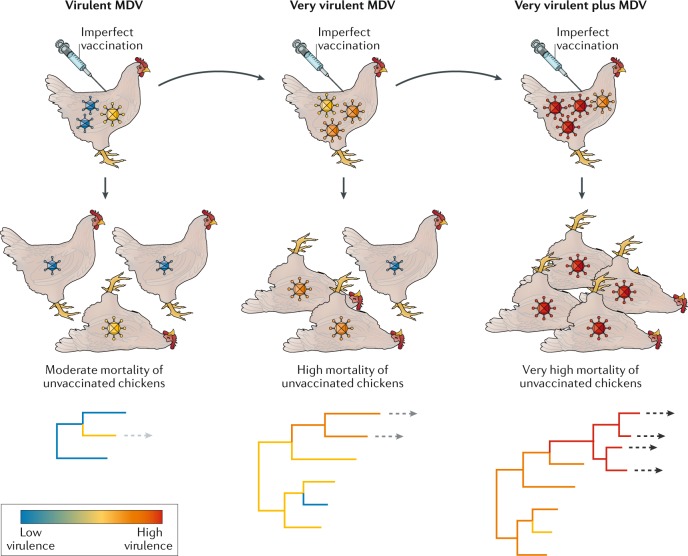
In the 1960s, a vaccine was developed for Marek’s disease virus (MDV) of chickens present on poultry farms. This imperfect vaccine reduced disease symptoms but did not prevent virus replication, thereby extending the infectious periods, and hence potential for transmission, of virulent strains that would have been removed by natural selection before transmission to a new host in the pre-vaccine era. Because of this, ‘very virulent’ MDV began to appear within 10 years, necessitating the development of a second-generation vaccine that was also imperfect. This was followed, in an even shorter period, by the appearance of ‘very virulent plus’ MDV, requiring a third-generation vaccine. Although the genomic basis of MDV virulence is currently unknown, the phylogenies at the bottom of the figure hypothetically assign virulence to multiple causative mutations (as in the case of myxoma virus). The dashed arrows indicate the evolution of viruses to the next virulence grade.
A model Ebola virus (EBOV; Makona variant) phylogeny illustrates the evolution of a single amino acid substitution (glycoprotein, A82V) that is associated with viral adaptation to the human host during the West African EBOV outbreak of 2013–2016. A82V improves binding to the human NPC1 receptor utilized by EBOV, increasing infectivity in humans (red) while simultaneously reducing infectivity in cells from the bat reservoir species (blue),. Maps above the phylogeny show the spread of EBOV over the timeline of the outbreak in the three affected countries in West Africa, where blue-shaded regions correspond to the wild-type virus variant (A82) and red-shaded areas correspond to mutated virus variant (V82). It is possible that A82V was also associated with an increase in both EBOV case numbers and mortality (that is, virulence) as the outbreak progressed, such that increased virulence is directly selectively advantageous, although this is confounded by epidemiological factors.
References
-
- Power RA, Parkhill J, de Oliveira T. Microbial genome-wide association studies: lessons from human GWAS. Nat. Rev. Genet. 2017;18:41–50. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
