

Molecular basis of cystic fibrosis: from bench to bedside
- PMID: 30306073
- PMCID: PMC6174194
- DOI: 10.21037/atm.2018.06.48
Molecular basis of cystic fibrosis: from bench to bedside
Abstract
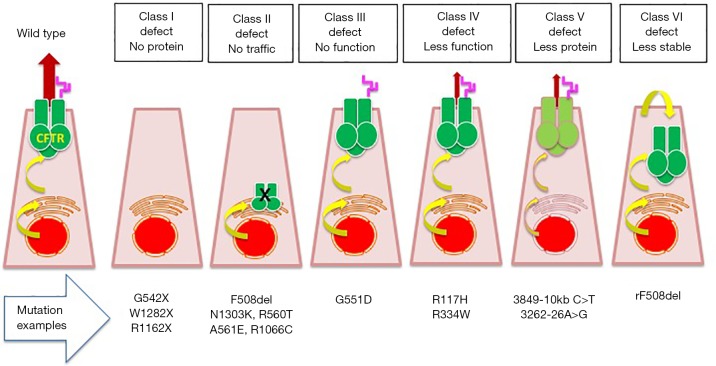
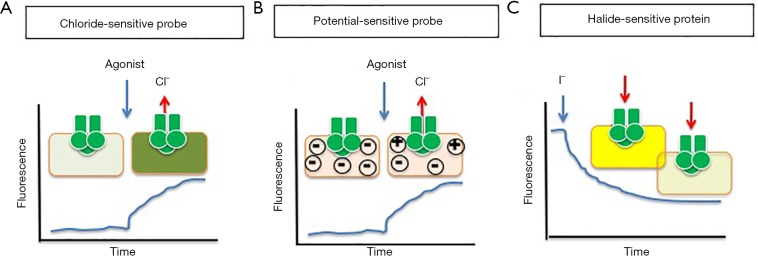
Cystic fibrosis (CF), is an autosomal recessive disease affecting different organs. The lung disease, characterized by recurrent and chronic bacterial infection and inflammation since infancy, is the main cause of morbidity and precocious mortality of these individuals. The innovative therapies directed to repair the defective CF gene should account for the presence of more than 200 disease-causing mutations of the CF transmembrane conductance regulator (CFTR) gene. The review will recall the different experimental approaches in discovering CFTR protein targeted molecules, such as the high throughput screening on chemical libraries to discover correctors and potentiators of CFTR protein, dual-acting compounds, read-through molecules, splicing defects repairing tools, CFTR "amplifiers".
Keywords: CFTR potentiators; Cystic fibrosis (CF); cystic fibrosis transmembrane conductance regulator correctors (CFTR correctors); personalized medicine.
Conflict of interest statement
Conflicts of Interest: The authors have no conflicts of interest to declare.
Figures
Similar articles
-
Innovative Therapies for Cystic Fibrosis: The Road from Treatment to Cure.Mol Diagn Ther. 2019 Apr;23(2):263-279. doi: 10.1007/s40291-018-0372-6. Mol Diagn Ther. 2019. PMID: 30478715 Review.
-
F508del-cystic fibrosis transmembrane regulator correctors for treatment of cystic fibrosis: a patent review.Expert Opin Ther Pat. 2015;25(9):991-1002. doi: 10.1517/13543776.2015.1045878. Epub 2015 May 15. Expert Opin Ther Pat. 2015. PMID: 25971311 Review.
-
Repairing mutated proteins--development of small molecules targeting defects in the cystic fibrosis transmembrane conductance regulator.Expert Opin Drug Discov. 2013 Jun;8(6):691-708. doi: 10.1517/17460441.2013.788495. Epub 2013 Apr 11. Expert Opin Drug Discov. 2013. PMID: 23574506 Review.
-
CFTR Modulators: The Changing Face of Cystic Fibrosis in the Era of Precision Medicine.Front Pharmacol. 2020 Feb 21;10:1662. doi: 10.3389/fphar.2019.01662. eCollection 2019. Front Pharmacol. 2020. PMID: 32153386 Free PMC article. Review.
-
Discovery and SAR of 4-aminopyrrolidine-2-carboxylic acid correctors of CFTR for the treatment of cystic fibrosis.Bioorg Med Chem Lett. 2022 Sep 15;72:128843. doi: 10.1016/j.bmcl.2022.128843. Epub 2022 Jun 7. Bioorg Med Chem Lett. 2022. PMID: 35688367
Cited by
-
Synthesis and Therapeutic Applications of Iminosugars in Cystic Fibrosis.Int J Mol Sci. 2020 May 9;21(9):3353. doi: 10.3390/ijms21093353. Int J Mol Sci. 2020. PMID: 32397443 Free PMC article. Review.
-
Membrane proteins enter the fold.Curr Opin Struct Biol. 2021 Aug;69:124-130. doi: 10.1016/j.sbi.2021.03.006. Epub 2021 May 8. Curr Opin Struct Biol. 2021. PMID: 33975156 Free PMC article. Review.
-
VX-445 (elexacaftor) inhibits chloride secretion across human bronchial epithelial cells by directly blocking KCa3.1 channels.PNAS Nexus. 2025 Jul 4;4(7):pgaf211. doi: 10.1093/pnasnexus/pgaf211. eCollection 2025 Jul. PNAS Nexus. 2025. PMID: 40688096 Free PMC article.
-
Recent Strategic Advances in CFTR Drug Discovery: An Overview.Int J Mol Sci. 2020 Mar 31;21(7):2407. doi: 10.3390/ijms21072407. Int J Mol Sci. 2020. PMID: 32244346 Free PMC article. Review.
-
Combined Treatment of Bronchial Epithelial Calu-3 Cells with Peptide Nucleic Acids Targeting miR-145-5p and miR-101-3p: Synergistic Enhancement of the Expression of the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Gene.Int J Mol Sci. 2022 Aug 19;23(16):9348. doi: 10.3390/ijms23169348. Int J Mol Sci. 2022. PMID: 36012615 Free PMC article.
References
Publication types
LinkOut - more resources
Full Text Sources