

The Janus face of HMGB1 in heart disease: a necessary update
- PMID: 30306212
- PMCID: PMC6339675
- DOI: 10.1007/s00018-018-2930-9
The Janus face of HMGB1 in heart disease: a necessary update
Abstract
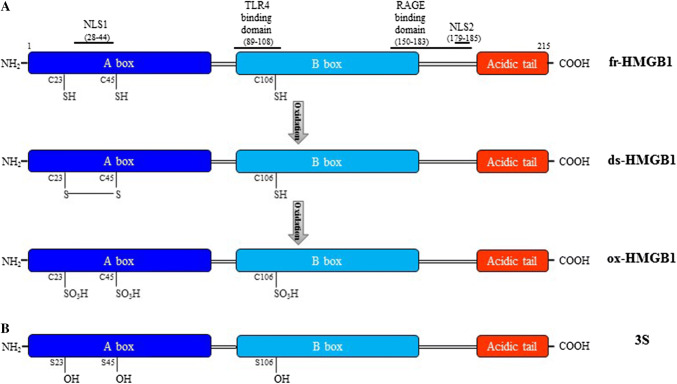
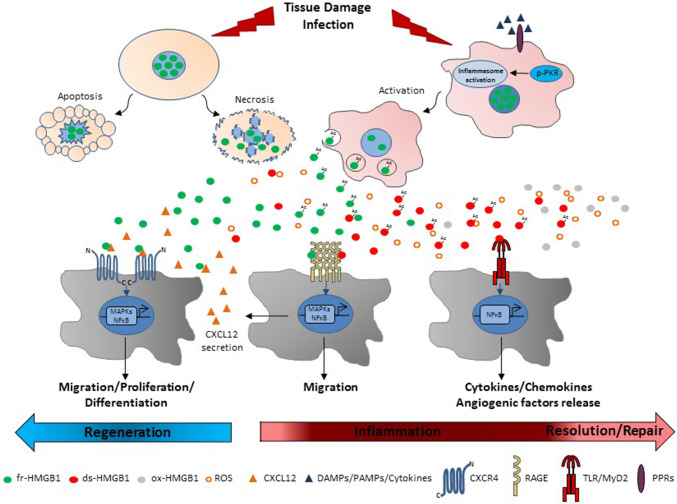
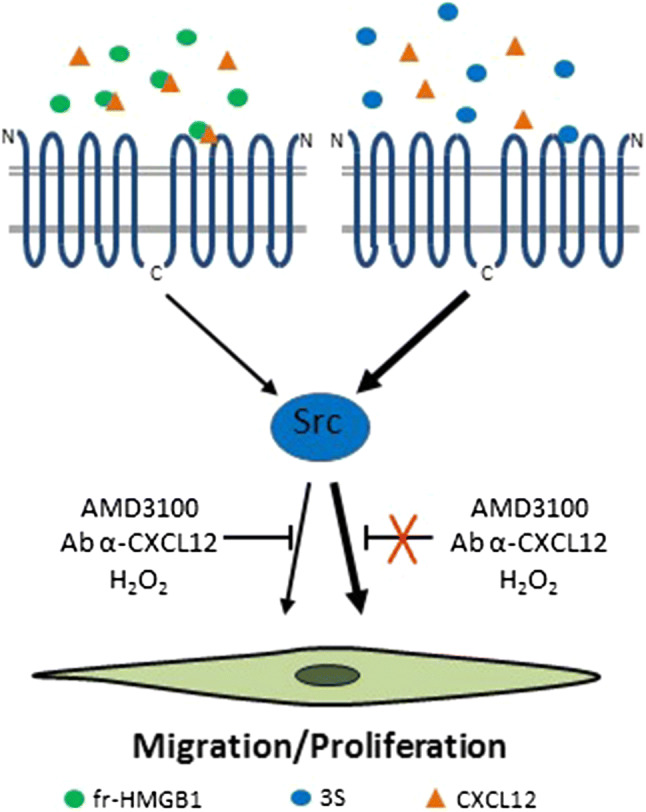
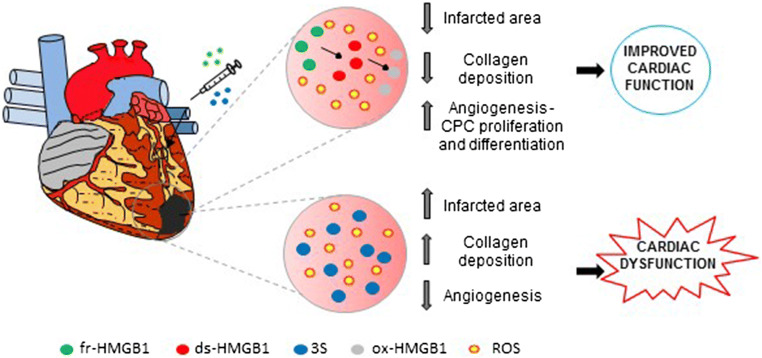
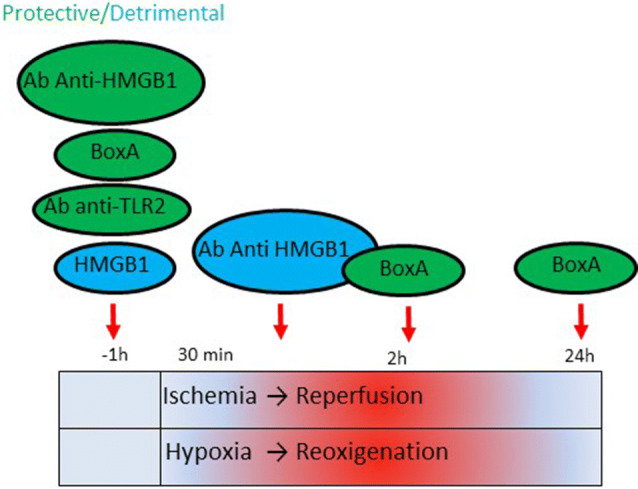
High mobility group box 1 (HMGB1) is a ubiquitous nuclear protein involved in transcription regulation, DNA replication and repair and nucleosome assembly. HMGB1 is passively released by necrotic tissues or actively secreted by stressed cells. Extracellular HMGB1 acts as a damage-associated molecular pattern (DAMPs) molecule and gives rise to several redox forms that by binding to different receptors and interactors promote a variety of cellular responses, including tissue inflammation or regeneration. Inhibition of extracellular HMGB1 in experimental models of myocardial ischemia/reperfusion injury, myocarditis, cardiomyopathies induced by mechanical stress, diabetes, bacterial infection or chemotherapeutic drugs reduces inflammation and is protective. In contrast, administration of HMGB1 after myocardial infarction induced by permanent coronary artery ligation ameliorates cardiac performance by promoting tissue regeneration. HMGB1 decreases contractility and induces hypertrophy and apoptosis in cardiomyocytes, stimulates cardiac fibroblast activities, and promotes cardiac stem cell proliferation and differentiation. Interestingly, maintenance of appropriate nuclear HMGB1 levels protects cardiomyocytes from apoptosis by preventing DNA oxidative stress, and mice with HMGB1cardiomyocyte-specific overexpression are partially protected from cardiac damage. Finally, higher levels of circulating HMGB1 are associated to human heart diseases. Hence, during cardiac injury, HMGB1 elicits both harmful and beneficial responses that may in part depend on the generation and stability of the diverse redox forms, whose specific functions in this context remain mostly unexplored. This review summarizes recent findings on HMGB1 biology and heart dysfunctions and discusses the therapeutic potential of modulating its expression, localization, and oxidative-dependent activities.
Keywords: Alarmin; Biomarker; Inflammation; Oxidative stress; Regeneration.
Conflict of interest statement
None declared. However, M.E.B. is founder and part owner of HMGBiotech, a company that provides goods and services related to HMGB proteins.
Figures

Similar articles
-
High Mobility Group Box 1 and Cardiovascular Diseases: Study of Act and Connect.Cardiovasc Toxicol. 2024 Nov;24(11):1268-1286. doi: 10.1007/s12012-024-09919-5. Epub 2024 Sep 6. Cardiovasc Toxicol. 2024. PMID: 39242448 Review.
-
HMGB1 and repair: focus on the heart.Pharmacol Ther. 2019 Apr;196:160-182. doi: 10.1016/j.pharmthera.2018.12.005. Epub 2018 Dec 6. Pharmacol Ther. 2019. PMID: 30529040 Review.
-
HMGB1-Mediated Activation of the Inflammatory-Reparative Response Following Myocardial Infarction.Cells. 2022 Jan 10;11(2):216. doi: 10.3390/cells11020216. Cells. 2022. PMID: 35053332 Free PMC article. Review.
-
Therapeutic potential of high mobility group box-1 in ischemic injury and tissue regeneration.Curr Vasc Pharmacol. 2011 Nov;9(6):677-81. doi: 10.2174/157016111797484125. Curr Vasc Pharmacol. 2011. PMID: 21692740 Review.
-
High Mobility Group Box 1: Biological Functions and Relevance in Oxidative Stress Related Chronic Diseases.Cells. 2022 Mar 1;11(5):849. doi: 10.3390/cells11050849. Cells. 2022. PMID: 35269471 Free PMC article. Review.
Cited by
-
The Trinity of cGAS, TLR9, and ALRs Guardians of the Cellular Galaxy Against Host-Derived Self-DNA.Front Immunol. 2021 Feb 11;11:624597. doi: 10.3389/fimmu.2020.624597. eCollection 2020. Front Immunol. 2021. PMID: 33643304 Free PMC article. Review.
-
Damage-associated molecular patterns in vitiligo: igniter fuse from oxidative stress to melanocyte loss.Redox Rep. 2022 Dec;27(1):193-199. doi: 10.1080/13510002.2022.2123864. Redox Rep. 2022. PMID: 36154894 Free PMC article. Review.
-
Lipopolysaccharides protect mesenchymal stem cell against cardiac ischemia-reperfusion injury by HMGB1/STAT3 signaling.J Geriatr Cardiol. 2023 Nov 28;20(11):801-812. doi: 10.26599/1671-5411.2023.11.007. J Geriatr Cardiol. 2023. PMID: 38098470 Free PMC article.
-
Correlation of HMGB1, PON-1, MCP-1, and Periodontal P. gingivalis with Amniotic Fluid Fecal Dye.J Healthc Eng. 2022 Feb 22;2022:3143102. doi: 10.1155/2022/3143102. eCollection 2022. J Healthc Eng. 2022. PMID: 35242296 Free PMC article.
-
Maslinic Acid Attenuates Ischemia/Reperfusion Injury-Induced Myocardial Inflammation and Apoptosis by Regulating HMGB1-TLR4 Axis.Front Cardiovasc Med. 2021 Nov 10;8:768947. doi: 10.3389/fcvm.2021.768947. eCollection 2021. Front Cardiovasc Med. 2021. PMID: 34859077 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
