

Acquired Resistance of MET-Amplified Non-small Cell Lung Cancer Cells to the MET Inhibitor Capmatinib
- PMID: 30309221
- PMCID: PMC6639226
- DOI: 10.4143/crt.2018.052
Acquired Resistance of MET-Amplified Non-small Cell Lung Cancer Cells to the MET Inhibitor Capmatinib
Abstract
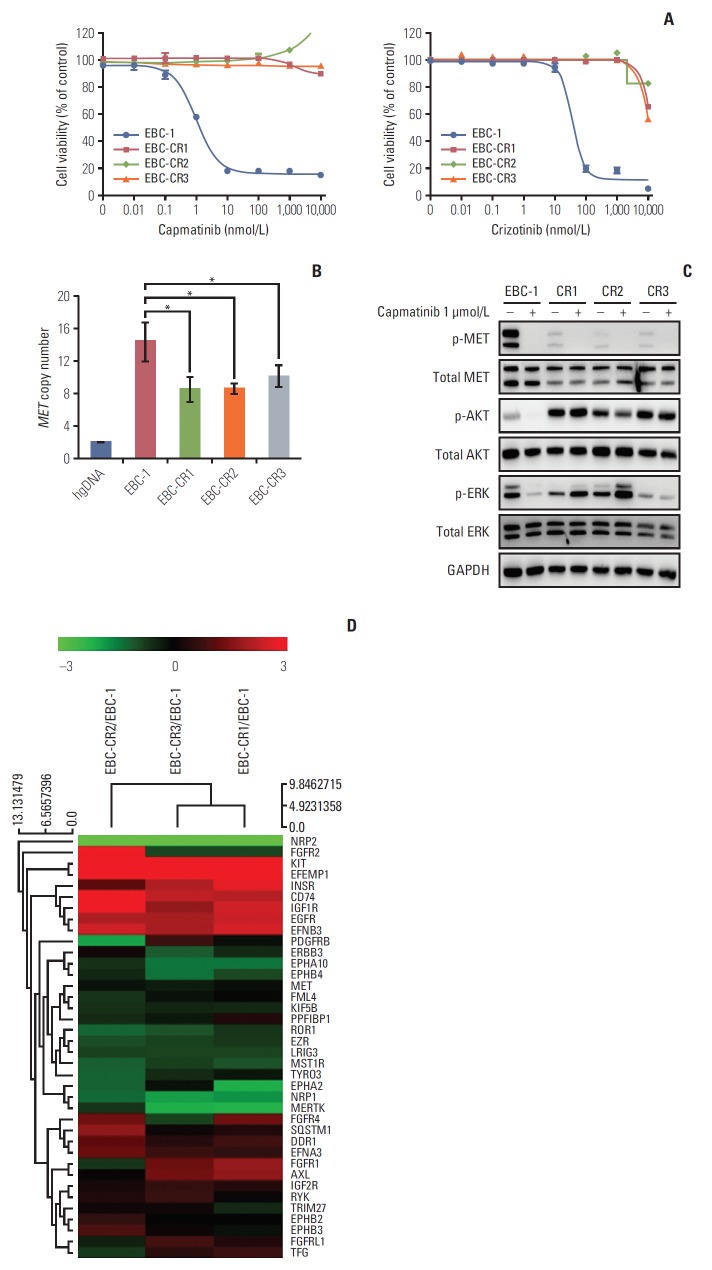
Purpose: Amplified mesenchymal-epithelial transition factor, MET, is a receptor tyrosine kinase (RTK) that has been considered a druggable target in non-small cell lung cancer (NSCLC). Although multiple MET tyrosine kinase inhibitors (TKIs) are being actively developed for MET-driven NSCLC, the mechanisms of acquired resistance to MET-TKIs have not been well elucidated. To understand the mechanisms of resistance and establish therapeutic strategies, we developed an in vitro model using the MET-amplified NSCLC cell line EBC-1.
Materials and methods: We established capmatinib-resistant NSCLC cell lines and identified alternative signaling pathways using 3' mRNA sequencing and human phospho-RTK arrays. Copy number alterations were evaluated by quantitative polymerase chain reaction and cell proliferation assay; activation of RTKs and downstream effectors were compared between the parental cell line EBC-1 and the resistant cell lines.
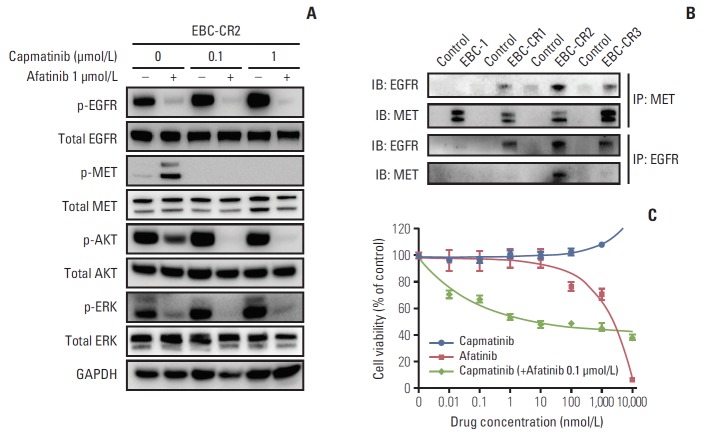
Results: We found that EBC-CR1 showed an epidermal growth factor receptor (EGFR)‒dependent growth and sensitivity to afatinib, an irreversible EGFR TKI. EBC-CR2 cells that had overexpression of EGFR-MET heterodimer dramatically responded to combined capmatinib with afatinib. In addition, EBC-CR3 cells derived from EBC-CR1 cells that activated EGFR with amplified phosphoinositide-3 kinase catalytic subunit α (PIK3CA) were sensitive to combined afatinib with BYL719, a phosphoinositide 3-kinase α (PI3Kα) inhibitor.
Conclusion: Our in vitro studies suggested that activation of EGFR signaling and/or genetic alteration of downstream effectors like PIK3CA were alternative resistance mechanisms used by capmatinib-resistant NSCLC cell lines. In addition, combined treatments with MET, EGFR, and PI3Kα inhibitors may be effective therapeutic strategies in capmatinib-resistant NSCLC patients.
Keywords: Acquired resistance; Capmatinib; MET amplification; MET tyrosine kinase inhibitor; Non-small cell lung carcinoma.
Conflict of interest statement
Conflict of interest relevant to this article was not reported.
Figures


Similar articles
-
Capmatinib for the treatment of non-small cell lung cancer.Expert Rev Anticancer Ther. 2019 Aug;19(8):659-671. doi: 10.1080/14737140.2019.1643239. Epub 2019 Aug 1. Expert Rev Anticancer Ther. 2019. PMID: 31368815 Review.
-
MET Gene Amplification and MET Receptor Activation Are Not Sufficient to Predict Efficacy of Combined MET and EGFR Inhibitors in EGFR TKI-Resistant NSCLC Cells.PLoS One. 2015 Nov 18;10(11):e0143333. doi: 10.1371/journal.pone.0143333. eCollection 2015. PLoS One. 2015. PMID: 26580964 Free PMC article.
-
Dual MET and ERBB inhibition overcomes intratumor plasticity in osimertinib-resistant-advanced non-small-cell lung cancer (NSCLC).Ann Oncol. 2017 Oct 1;28(10):2451-2457. doi: 10.1093/annonc/mdx396. Ann Oncol. 2017. PMID: 28961841 Free PMC article.
-
Phase Ib/II Study of Capmatinib (INC280) Plus Gefitinib After Failure of Epidermal Growth Factor Receptor (EGFR) Inhibitor Therapy in Patients With EGFR-Mutated, MET Factor-Dysregulated Non-Small-Cell Lung Cancer.J Clin Oncol. 2018 Nov 1;36(31):3101-3109. doi: 10.1200/JCO.2018.77.7326. Epub 2018 Aug 29. J Clin Oncol. 2018. PMID: 30156984 Clinical Trial.
-
Current mechanism of acquired resistance to epidermal growth factor receptor-tyrosine kinase inhibitors and updated therapy strategies in human nonsmall cell lung cancer.J Cancer Res Ther. 2016 Dec;12(Supplement):C131-C137. doi: 10.4103/0973-1482.200613. J Cancer Res Ther. 2016. PMID: 28230005 Review.
Cited by
-
Protein tyrosine kinase inhibitor resistance in malignant tumors: molecular mechanisms and future perspective.Signal Transduct Target Ther. 2022 Sep 17;7(1):329. doi: 10.1038/s41392-022-01168-8. Signal Transduct Target Ther. 2022. PMID: 36115852 Free PMC article. Review.
-
MYC amplification-conferred primary resistance to capmatinib in a MET-amplified NSCLC patient: a case report.Transl Lung Cancer Res. 2022 Sep;11(9):1967-1972. doi: 10.21037/tlcr-22-176. Transl Lung Cancer Res. 2022. PMID: 36248327 Free PMC article.
-
Non-small cell lung carcinoma (NSCLC): Implications on molecular pathology and advances in early diagnostics and therapeutics.Genes Dis. 2022 Aug 23;10(3):960-989. doi: 10.1016/j.gendis.2022.07.023. eCollection 2023 May. Genes Dis. 2022. PMID: 37396553 Free PMC article. Review.
-
SOS2 modulates the threshold of EGFR signaling to regulate osimertinib efficacy and resistance in lung adenocarcinoma.Mol Oncol. 2024 Mar;18(3):641-661. doi: 10.1002/1878-0261.13564. Epub 2024 Jan 18. Mol Oncol. 2024. PMID: 38073064 Free PMC article.
-
Novel Therapies for Metastatic Non-Small Cell Lung Cancer with MET Exon 14 Alterations: A Spotlight on Capmatinib.Lung Cancer (Auckl). 2021 Mar 18;12:11-20. doi: 10.2147/LCTT.S263610. eCollection 2021. Lung Cancer (Auckl). 2021. PMID: 33776501 Free PMC article. Review.
References
-
- Gschwind A, Fischer OM, Ullrich A. The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat Rev Cancer. 2004;4:361–70. - PubMed
-
- Schafer M, Werner S. Cancer as an overhealing wound: an old hypothesis revisited. Nat Rev Mol Cell Biol. 2008;9:628–38. - PubMed
-
- Jo M, Stolz DB, Esplen JE, Dorko K, Michalopoulos GK, Strom SC. Cross-talk between epidermal growth factor receptor and c-Met signal pathways in transformed cells. J Biol Chem. 2000;275:8806–11. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
