

Towards a Quantitative Understanding of Cell Identity
- PMID: 30309735
- PMCID: PMC6249108
- DOI: 10.1016/j.tcb.2018.09.002
Towards a Quantitative Understanding of Cell Identity
Abstract
Cells have traditionally been characterized using expression levels of a few proteins that are thought to specify phenotype. This requires a priori selection of proteins, which can introduce descriptor bias, and neglects the wealth of additional molecular information nested within each cell in a population, which often makes these sparse descriptors qualitative. Recently, more unbiased and quantitative cell characterization has been made possible by new high-throughput, information-dense experimental approaches and data-driven computational methods. This review discusses such quantitative descriptors in the context of three central concepts of cell identity: definition, creation, and stability. Collectively, these concepts are essential for constructing quantitative phenotypic landscapes, which will enhance our understanding of cell biology and facilitate cell engineering for research and clinical applications.
Keywords: cell phenotype; cellular decision making; computational modeling; high-throughput data analysis; network biology; phenotypic landscape.
Copyright © 2018 Elsevier Ltd. All rights reserved.
Figures
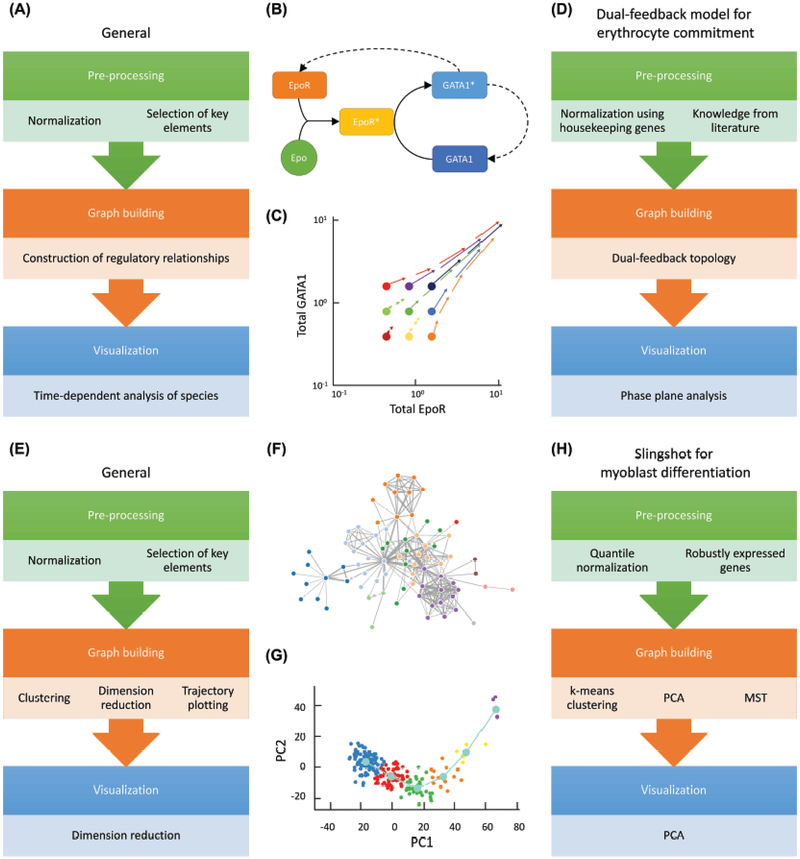
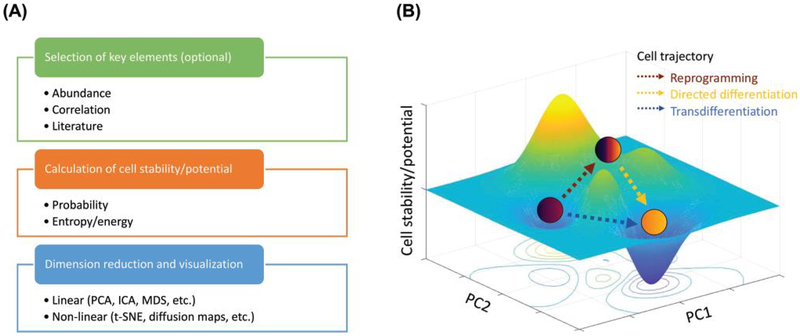
References
-
- Mazzarello P A unifying concept: the history of cell theory. Nat Cell Biol. 1999;1:E13. - PubMed
-
- Gall JG, McIntosh JR, editors. Landmark Papers in Cell Biology: Selected Research Articles Celebrating Forty Years of The American Society for Cell Biology. Cold Spring Harbor, NY : Bethesda, MD: : American Society for Cell Biology: Cold Spring Harbor Laboratory Pr; 2000.
-
- Schmidl C, Hansmann L, Lassmann T, Balwierz PJ, Kawaji H, Itoh M, et al. The enhancer and promoter landscape of human regulatory and conventional T-cell subpopulations. Blood. 2014;123:e68–78. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
