

Selective loss of function variants in IL6ST cause Hyper-IgE syndrome with distinct impairments of T-cell phenotype and function
- PMID: 30309848
- PMCID: PMC6395342
- DOI: 10.3324/haematol.2018.194233
Selective loss of function variants in IL6ST cause Hyper-IgE syndrome with distinct impairments of T-cell phenotype and function
Abstract
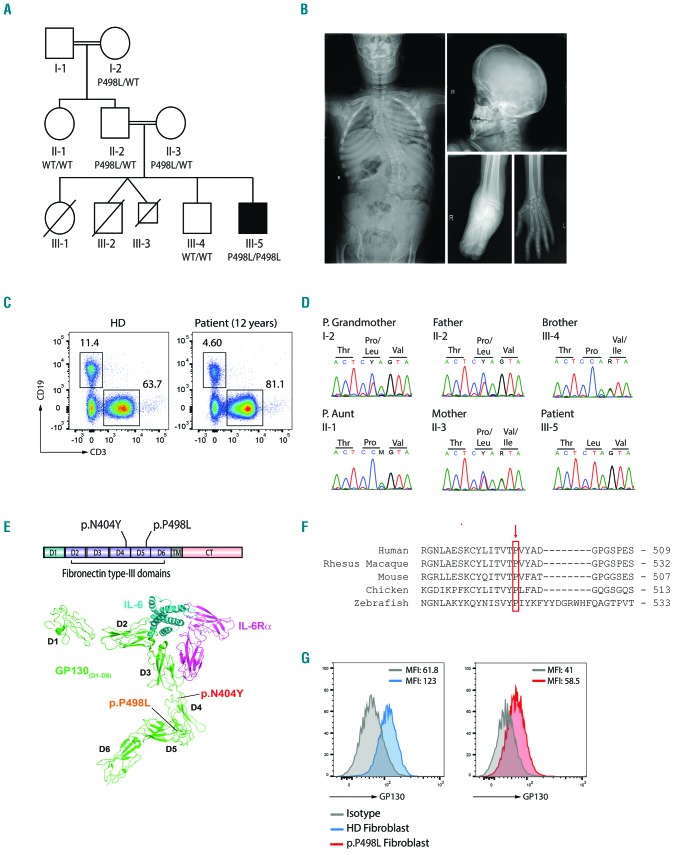
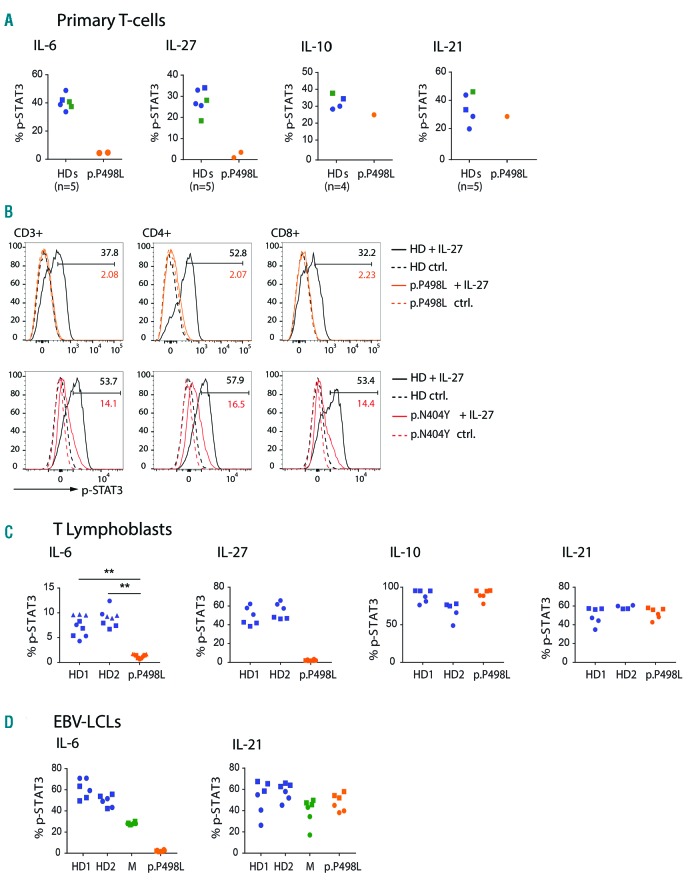
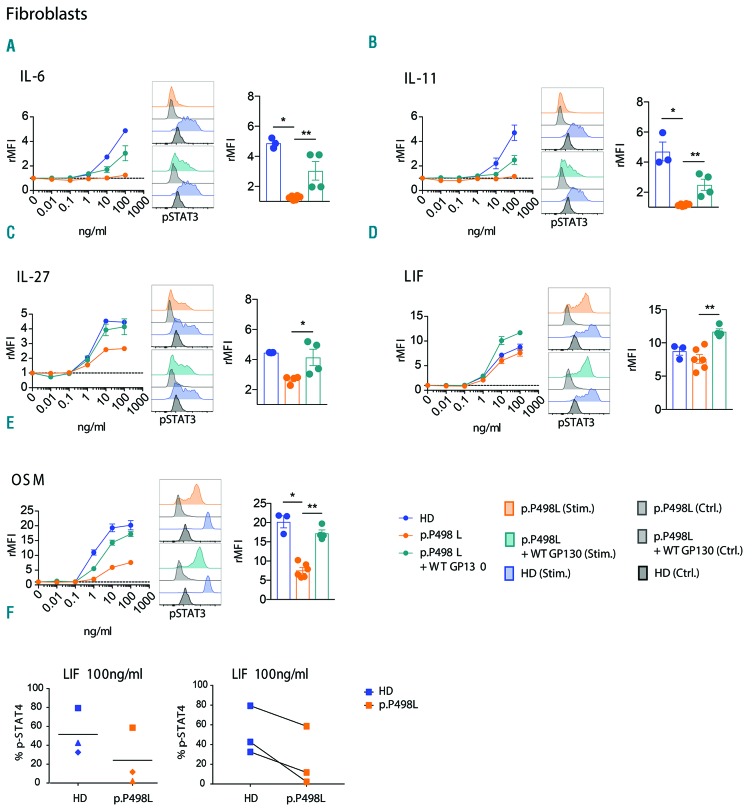
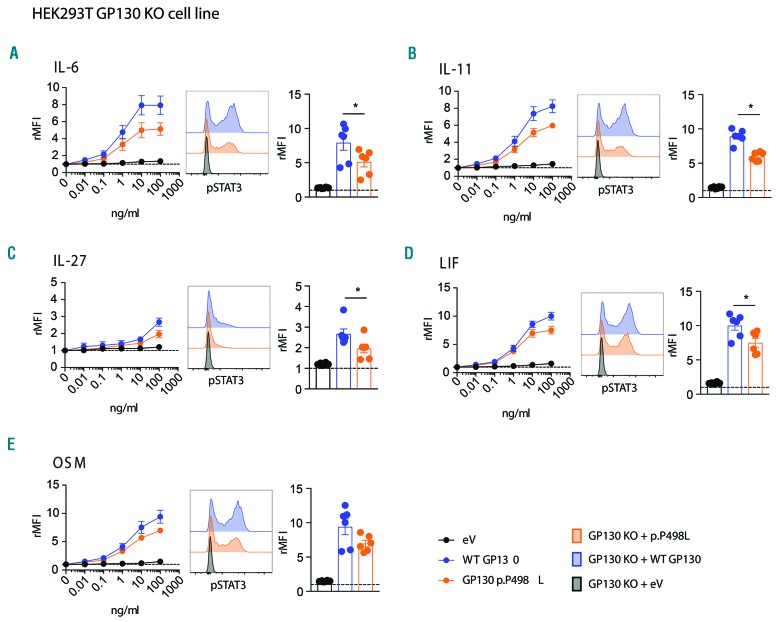
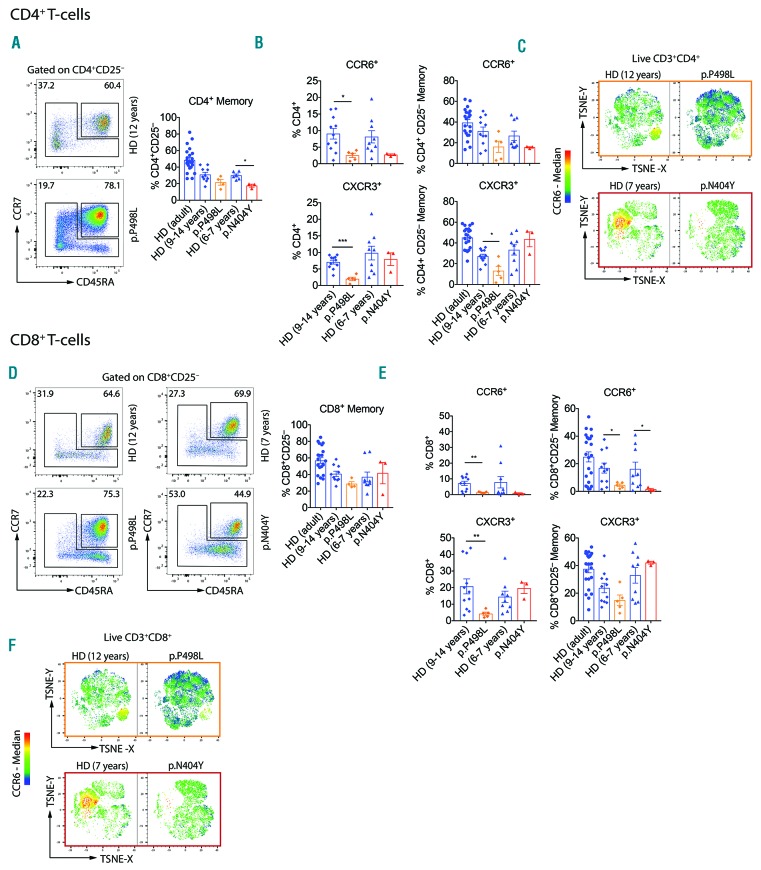
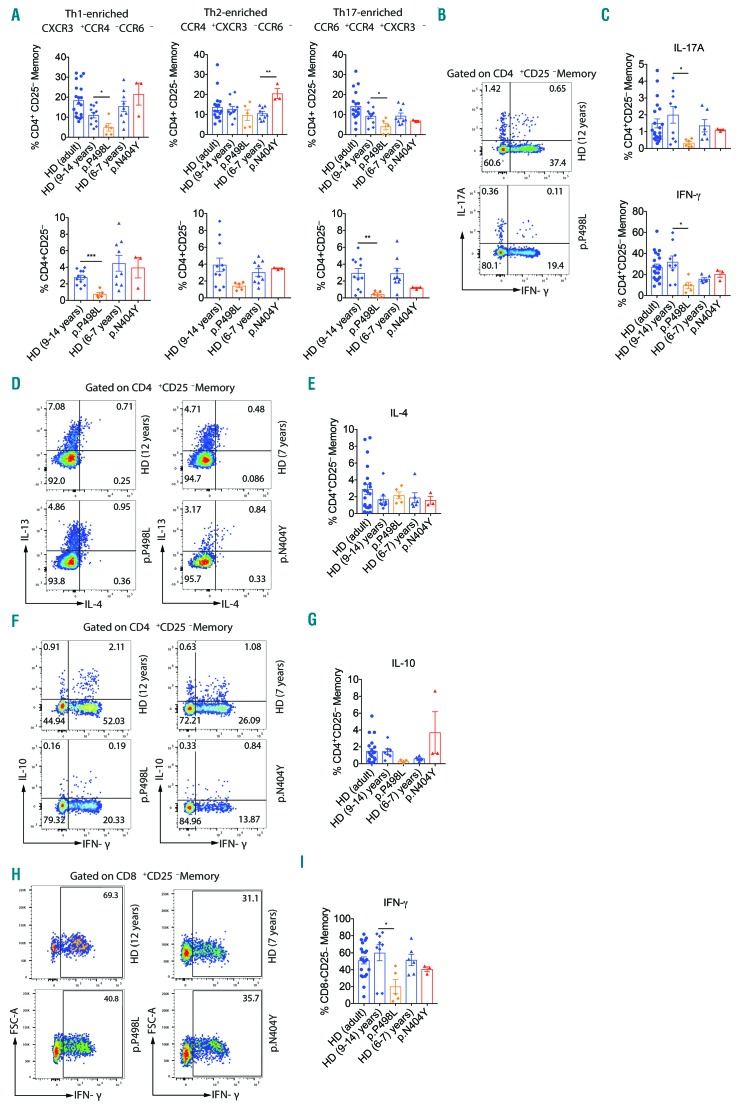
Hyper-IgE syndromes comprise a group of inborn errors of immunity. STAT3-deficient hyper-IgE syndrome is characterized by elevated serum IgE levels, recurrent infections and eczema, and characteristic skeletal anomalies. A loss-of-function biallelic mutation in IL6ST encoding the GP130 receptor subunit (p.N404Y) has very recently been identified in a singleton patient (herein referred to as PN404Y) as a novel etiology of hyper-IgE syndrome. Here, we studied a patient with hyper-IgE syndrome caused by a novel homozygous mutation in IL6ST (p.P498L; patient herein referred to as PP498L) leading to abrogated GP130 signaling after stimulation with IL-6 and IL-27 in peripheral blood mononuclear cells as well as IL-6 and IL-11 in fibroblasts. Extending the initial identification of selective GP130 deficiency, we aimed to dissect the effects of aberrant cytokine signaling on T-helper cell differentiation in both patients. Our results reveal the importance of IL-6 signaling for the development of CCR6-expressing memory CD4+ T cells (including T-helper 17-enriched subsets) and non-conventional CD8+T cells which were reduced in both patients. Downstream functional analysis of the GP130 mutants (p.N404Y and p.P498L) have shown differences in response to IL-27, with the p.P498L mutation having a more severe effect that is reflected by reduced T-helper 1 cells in this patient (PP498L) only. Collectively, our data suggest that characteristic features of GP130-deficient hyper-IgE syndrome phenotype are IL-6 and IL-11 dominated, and indicate selective roles of aberrant IL-6 and IL-27 signaling on the differentiation of T-cell subsets.
Copyright© 2019 Ferrata Storti Foundation.
Figures
References
-
- Grimbacher B, Holland SM, Gallin JI, et al. Hyper-IgE Syndrome with Recurrent Infections — An Autosomal Dominant Multisystem Disorder. N Engl J Med. 1999; 340(9):692–702. - PubMed
-
- Grimbacher B, Holland SM, Puck JM. Hyper-IgE syndromes. Immunol Rev. 2005; 203(1):244–250. - PubMed
-
- Holland SM, DeLeo FR, Elloumi HZ, et al. STAT3 Mutations in the Hyper-IgE Syndrome. N Engl J Med. 2007;357(16): 1608–1619. - PubMed
-
- Minegishi Y, Saito M, Tsuchiya S, et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature. 2007;448(7157):1058–1062. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
