

Review
doi: 10.1039/c8np00075a.
The expanding world of biosynthetic pericyclases: cooperation of experiment and theory for discovery
Affiliations
- PMID: 30311924
- PMCID: PMC6461539
- DOI: 10.1039/c8np00075a
Item in Clipboard
Review
The expanding world of biosynthetic pericyclases: cooperation of experiment and theory for discovery
Nat Prod Rep.
.
Abstract
Covering: 2000 to 2018 Pericyclic reactions are a distinct class of reactions that have wide synthetic utility. Before the recent discoveries described in this review, enzyme-catalyzed pericyclic reactions were not widely known to be involved in biosynthesis. This situation is changing rapidly. We define the scope of pericyclic reactions, give a historical account of their discoveries as biosynthetic reactions, and provide evidence that there are many enzymes in nature that catalyze pericyclic reactions. These enzymes, the "pericyclases," are the subject of this review.
Conflict of interest statement
Conflicts of interest
There are no conflicts to declare.
Figures
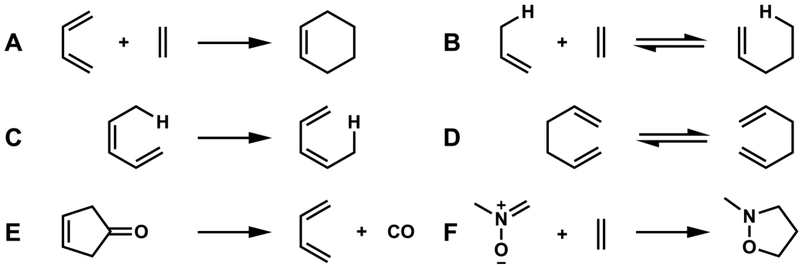
. A. Cycloaddition (Diels-Alder reaction). B. Alder-ene reaction (a Miscellaneozation). C. 1,5-Sigmatropic hydrogen shift, D. [3,3]-Sigmatropic shift (Cope rearrangement). E. Cheletropic reaction. F. A 1,3-dipolar cycloaddition, a hetero-pericyclic reaction.
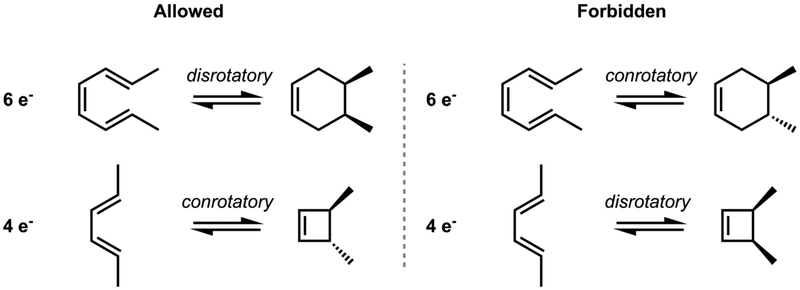
Allowed and forbidden electrocyclic reactions involving 6 and 4 electrons. In each case, only the allowed pathways are observed. Conrotatory, both termini rotate in the same direction; disrotatory, two termini rotate in the opposite direction.
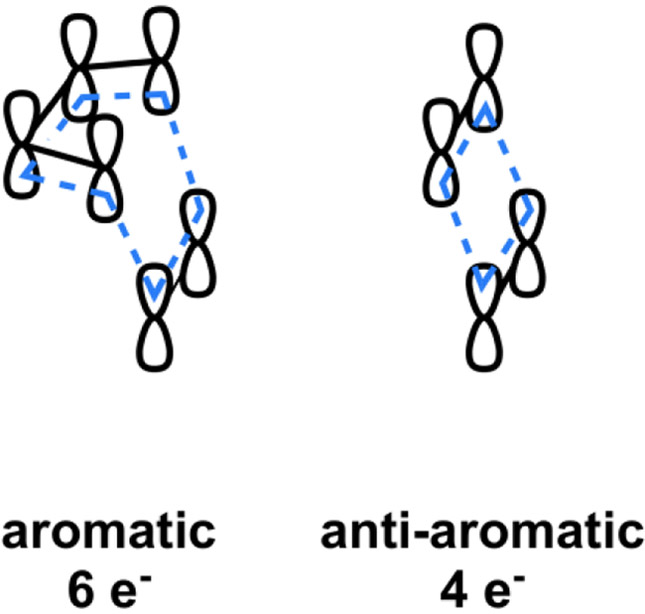
Schematic representation of the aromaticity of the allowed Diels-Alder transition state and anti-aromaticity of the [2+2] cycloaddition transition state.
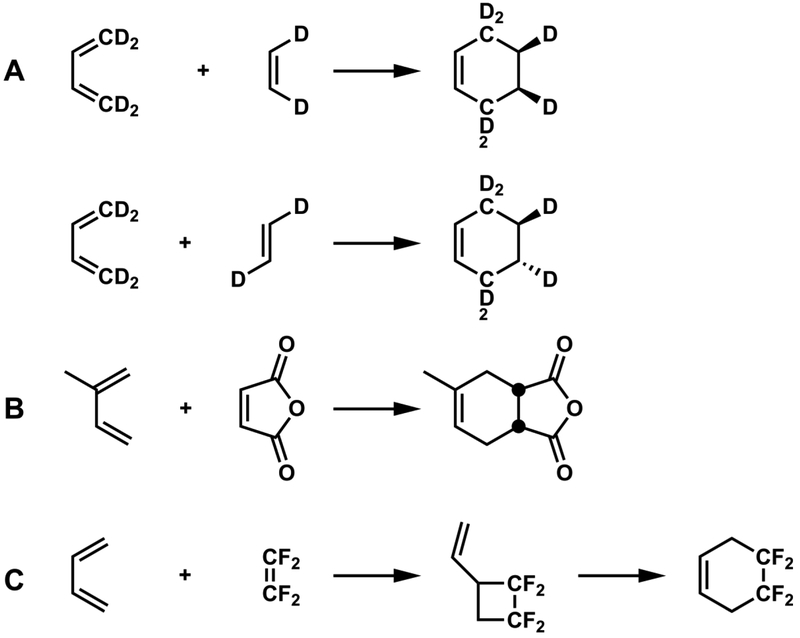
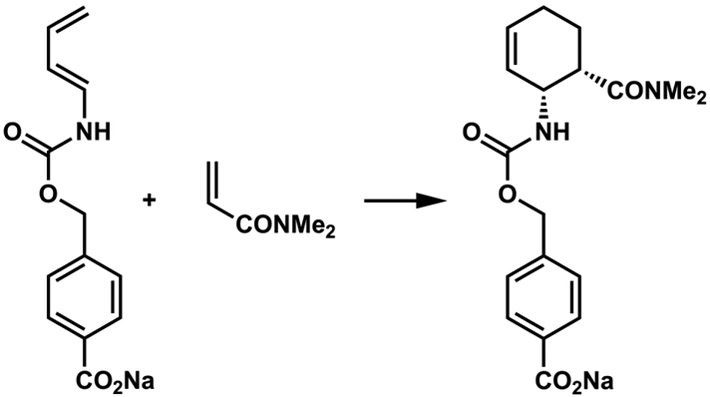
Diels-Alder reactions: Concerted proven by (A) stereospecificity3 and (B) kinetic isotope effects.4 Stepwise proven by (C) identification of an intermediate cyclobutane.
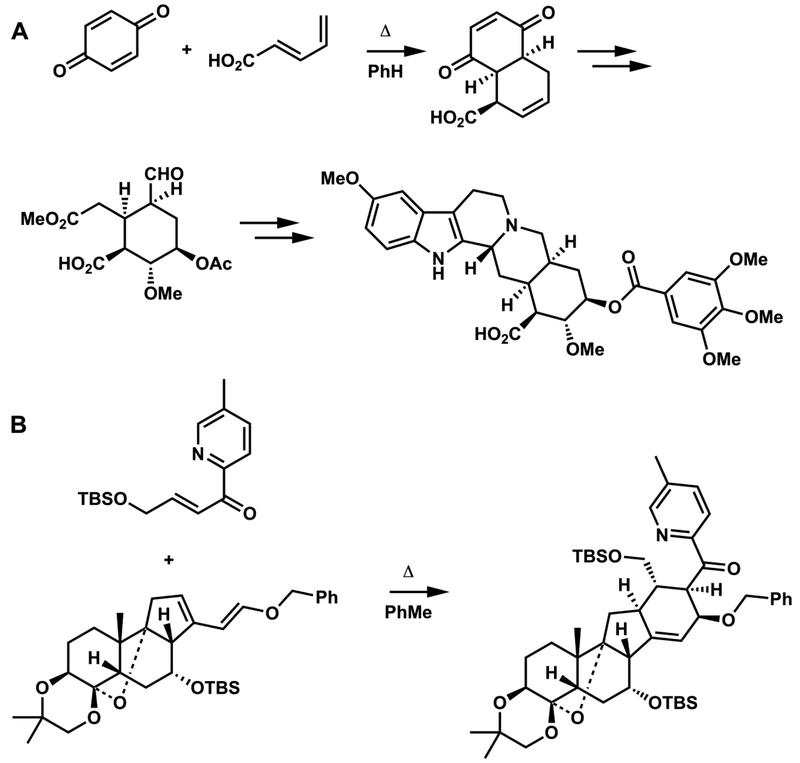
Diels-Alder steps in (A) Woodward’s reserpine synthesis (1956) and (B) Stork’s approach to the synthesis of 4-methylenegermine (2017).
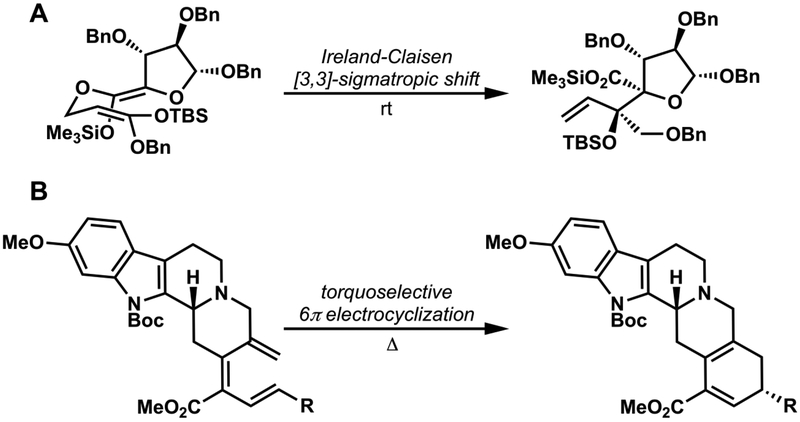
Examples of (A) a [3,3]-sigmatropic shift11 and (B) an electrocyclization12 involved in synthesis.
The Diels-Alder reaction catalyzed by a catalytic antibody17 and later by a computationally designed enzyme.
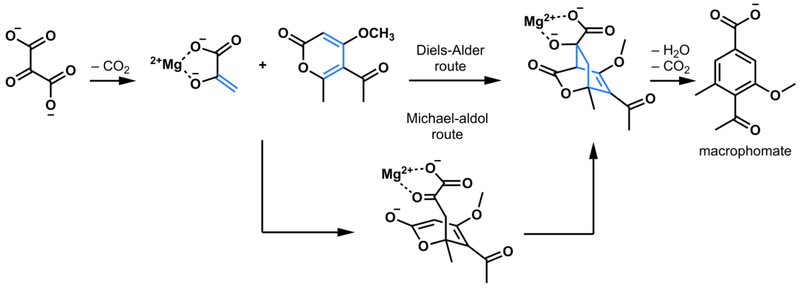
Reaction scheme comparing the concerted and stepwise Diels-Alder route to macrophomate catalyzed by macrophomate synthase
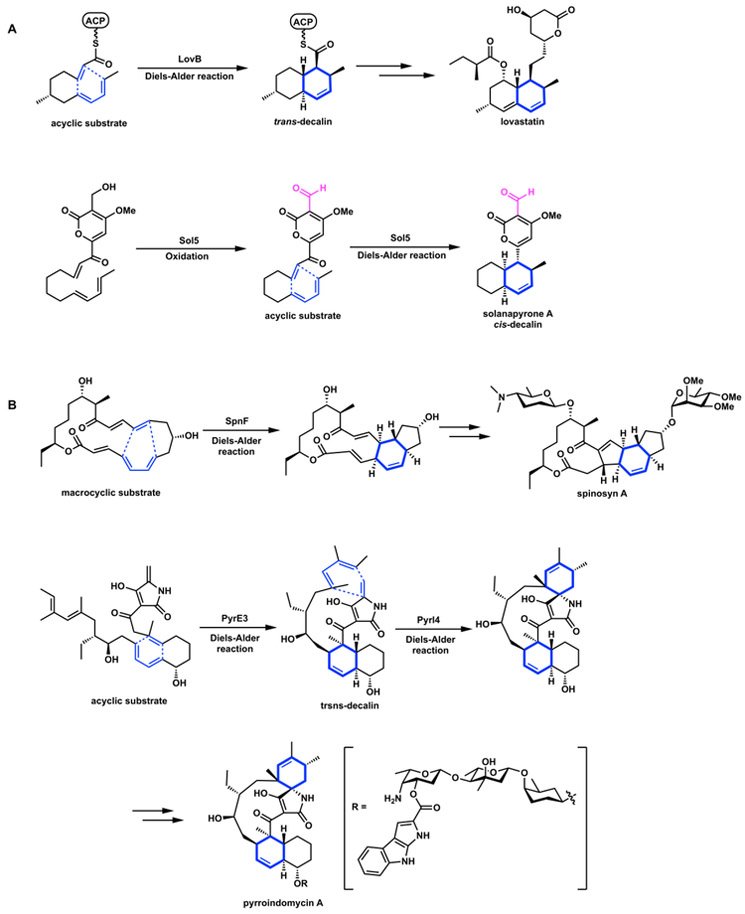
Examples of enzyme-catalyzed Diels-Alder reaction. (A) Multifunctional enzymes that have Diels-Alderases activities, LovB and Sol5. (B) Stand-alone Diels-Alderases, SpnF from spinosyn A biosynthesis, and PyrE3 and Pyl4 from pyrroindomycin A biosynthesis.
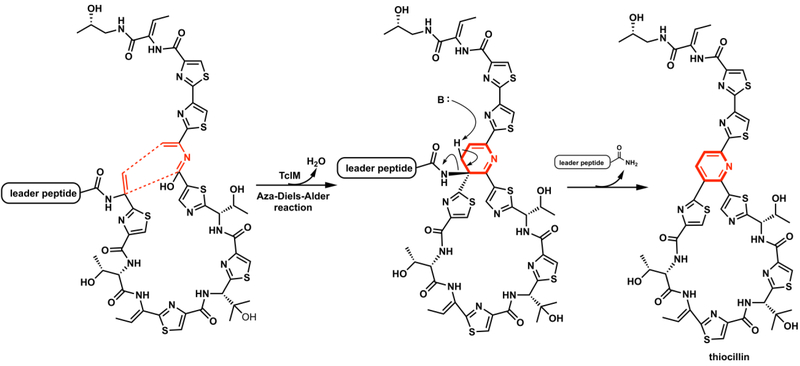
Enzymatic intramolecular hetero Diels–Alder reaction in formation of thiocillin.
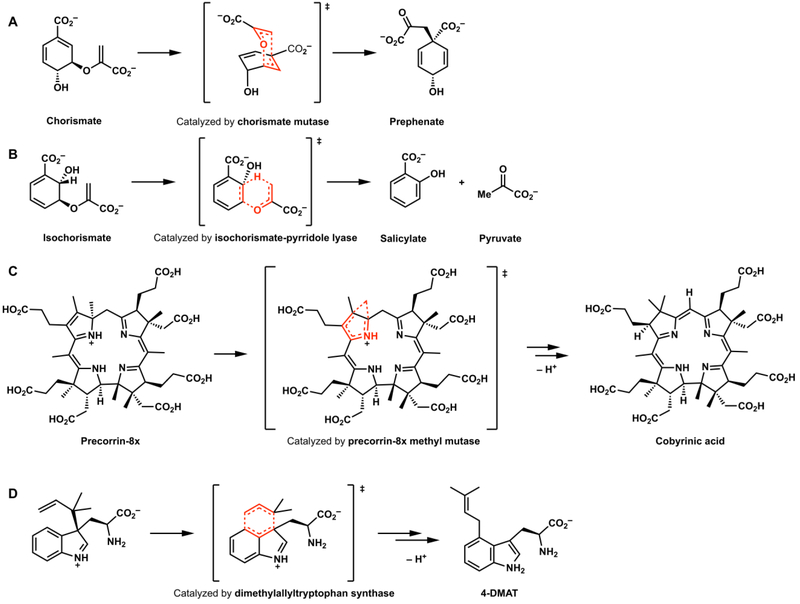
A. The Claisen rearrangement ([3,3]-sigmatropic shift) catalyzed by chorismate mutase; reactions putatively catalyzed by (B) isochorismate-pyrridole lyase, (C) precorrin-8x methyl mutase, and (D) dimethylallyltryptophane synthase.
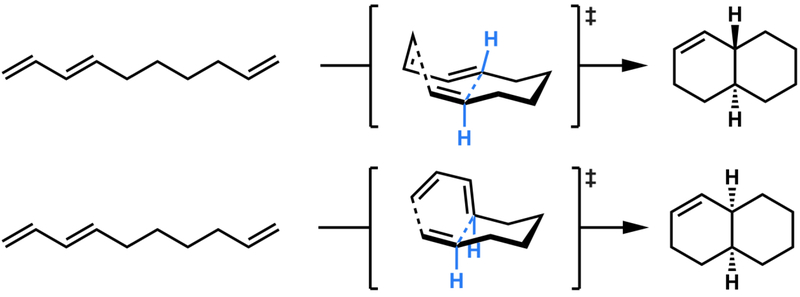
Intramolecular Diels-Alder reactions to form cis- and trans-octahydrodecalins.
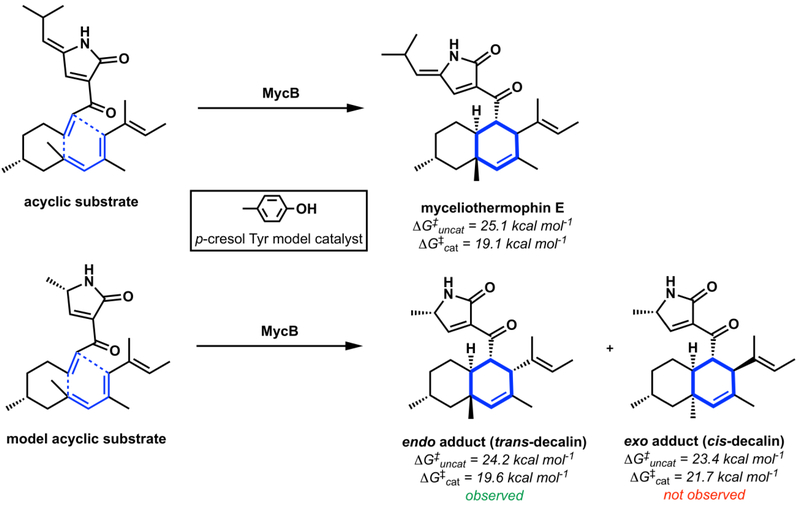
The MycB-catalyzed Diels-Alder reaction with calculated free energy barriers for spontaneous and model-catalyzed reactions.
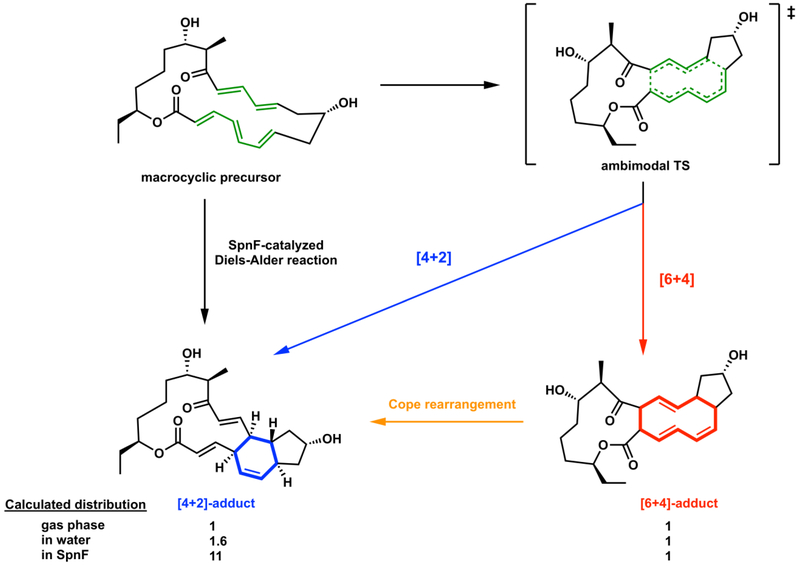
SpnF-catalyzed transannular cycloaddition reactions of the macrocyclic precursor via the single ambimodal transition state to form the [4+2] and [6+4] adducts. Ratios given are from MD simulations, although the rapid Cope rearrangement converts the [6+4] adduct to the more stable Diels-Alder adduct for thermodynamic reasons.
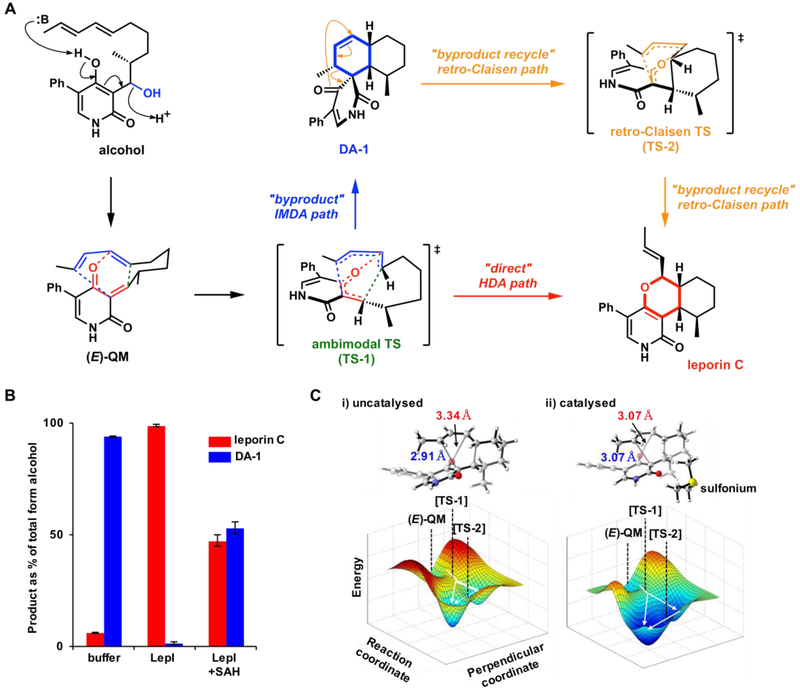
A. Reactions catalyzed by LepI in leporin C biosynthesis. B. Summary of cascade of LepI-catalyzed reactions. C. Ambimodal transition state structures and asymmetrical bifurcating PES for the formation of leporin C and DA-1 from (E)-QM.
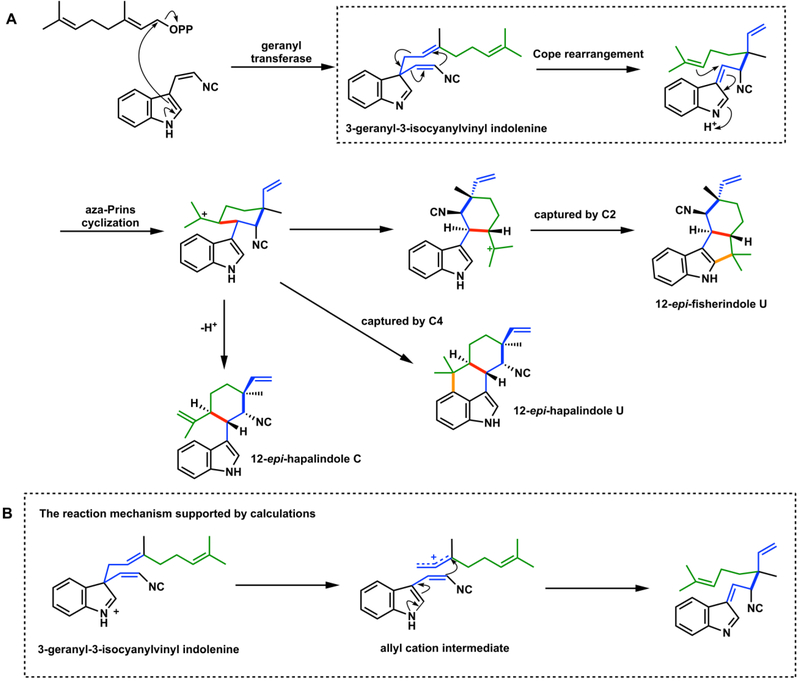
A. The Cope rearrangement involved in hapalindole and fischerindole biosynthesis. B. The likely acid-catalyzed stepwise (nonpericyclic) mechanism.,
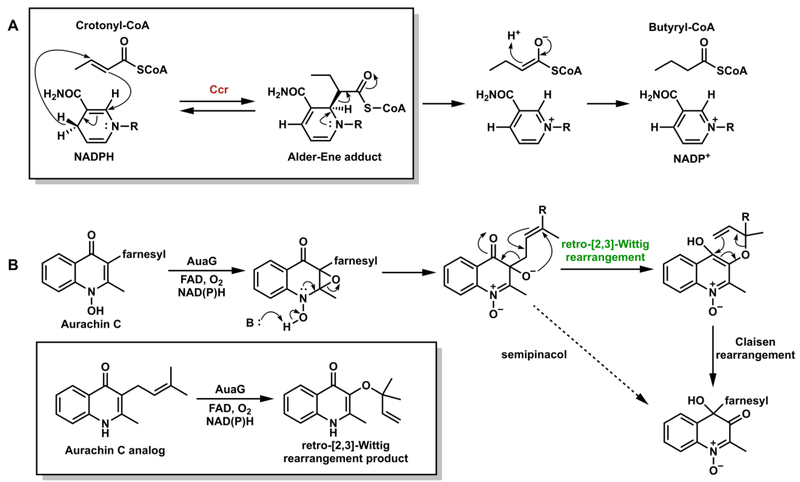
Proposed pericyclic (A) Alder-ene reaction in catalysis of Ccr;84 (B) retro-[2,3]-Wittig and Claisen rearrangements catalyzed by AuaG.
References
-
- Hoffmann R and Woodward RB, Acc. Chem. Res, 1968, 1, 17–22.
- Woodward RBand Hoffmann R, Angew. Chem. Int. Ed. Engl 1969, 8, 781; Verlag Chemie BmgH, Academic Press, 1970.
-
-
Ref. 1, page 169.
-
-
- Houk KN, Lin Y-T, and Brown FK, J. Am. Chem. Soc, 1986, 108, 554. - PubMed
-
- Beno BR, Houk KN, and Singleton DA, J. Am. Chem. Soc, 1996, 118, 9984–9985.
-
- Getty SJ and Borden WT, J. Am. Chem. Soc, 113, 4334–4335 (1991).