

A New Multisystem Disorder Caused by the Gαs Mutation p.F376V
- PMID: 30312418
- PMCID: PMC6380466
- DOI: 10.1210/jc.2018-01250
A New Multisystem Disorder Caused by the Gαs Mutation p.F376V
Abstract
Context: The α subunit of the stimulatory G protein (Gαs) links numerous receptors to adenylyl cyclase. Gαs, encoded by GNAS, is expressed predominantly from the maternal allele in certain tissues. Thus, maternal heterozygous loss-of-function mutations cause hormonal resistance, as in pseudohypoparathyroidism type Ia, whereas somatic gain-of-function mutations cause hormone-independent endocrine stimulation, as in McCune-Albright syndrome.
Objective: We report two unrelated boys presenting with a new combination of clinical findings that suggest both gain and loss of Gαs function.
Design and setting: Clinical features were studied and sequencing of GNAS was performed. Signaling capacities of wild-type and mutant Gαs were determined in the presence of different G protein-coupled receptors (GPCRs) under basal and agonist-stimulated conditions.
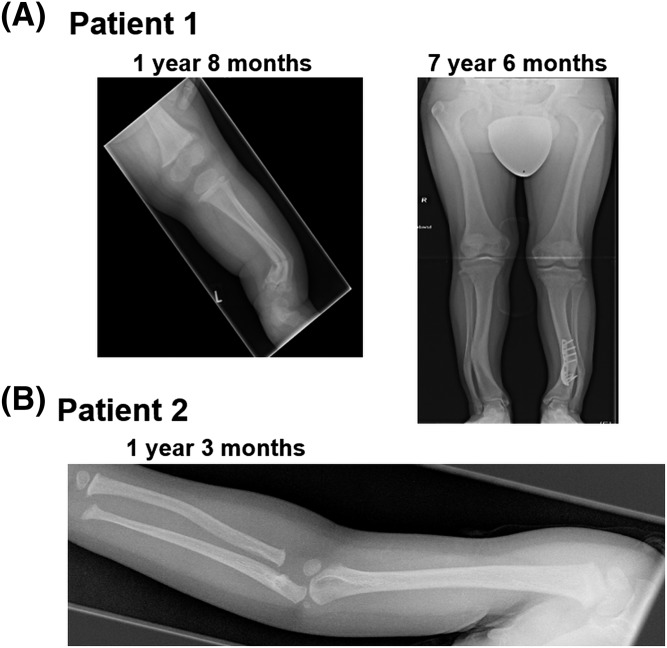
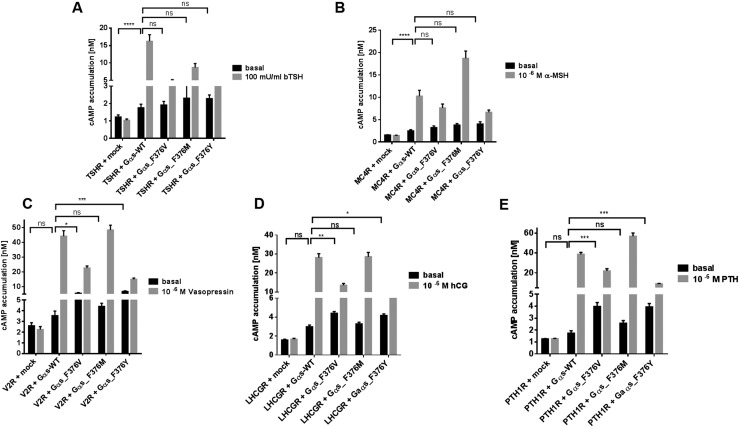
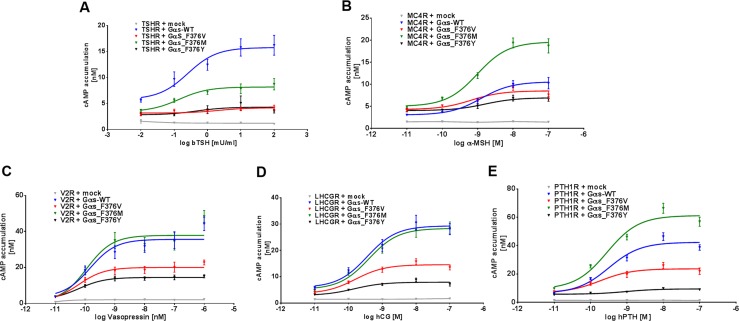
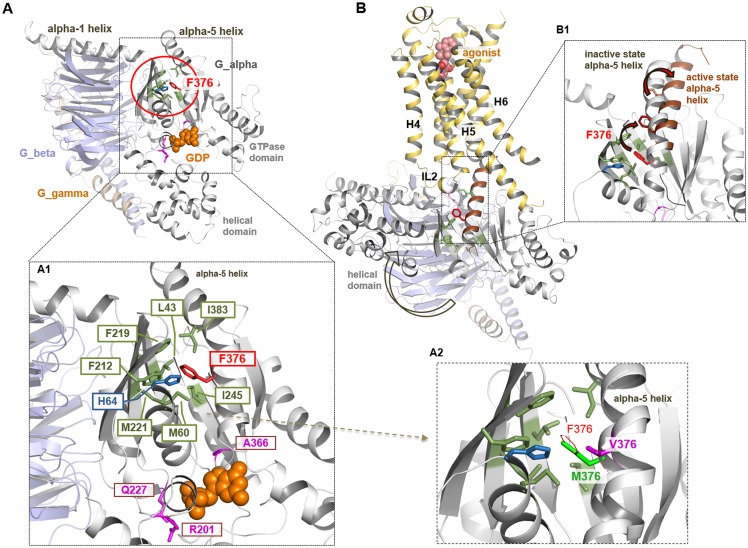
Results: Both unrelated patients presented with unexplained hyponatremia in infancy, followed by severe early onset gonadotrophin-independent precocious puberty and skeletal abnormalities. An identical heterozygous de novo variant (c.1136T>G; p.F376V) was found on the maternal GNAS allele in both patients; this resulted in a clinical phenotype that differed from known Gαs-related diseases and suggested gain of function at the vasopressin 2 receptor (V2R) and lutropin/choriogonadotropin receptor (LHCGR), yet increased serum PTH concentrations indicative of impaired proximal tubular PTH1 receptor (PTH1R) function. In vitro studies demonstrated that Gαs-F376V enhanced ligand-independent signaling at the PTH1R, LHCGR, and V2R and, at the same time, blunted ligand-dependent responses. Structural homology modeling suggested mutation-induced modifications at the C-terminal α5 helix of Gαs that are relevant for interaction with GPCRs and signal transduction.
Conclusions: The Gαs p.F376V mutation causes a previously unrecognized multisystem disorder.
Copyright © 2019 Endocrine Society.
Figures
References
-
- Oldham WM, Van Eps N, Preininger AM, Hubbell WL, Hamm HE. Mechanism of the receptor-catalyzed activation of heterotrimeric G proteins. Nat Struct Mol Biol. 2006;13(9):772–777. - PubMed
-
- Limbird LE. The receptor concept: a continuing evolution. Mol Interv. 2004;4(6):326–336. - PubMed
-
- Farfel Z, Bourne HR, Iiri T. The expanding spectrum of G protein diseases. N Engl J Med. 1999;340(13):1012–1020. - PubMed
-
- Spiegel AM, Weinstein LS. Inherited diseases involving G proteins and G protein-coupled receptors. Annu Rev Med. 2004;55(1):27–39. - PubMed
