

Spt6 Is Required for the Fidelity of Promoter Selection
- PMID: 30318445
- PMCID: PMC6239972
- DOI: 10.1016/j.molcel.2018.09.005
Spt6 Is Required for the Fidelity of Promoter Selection
Abstract
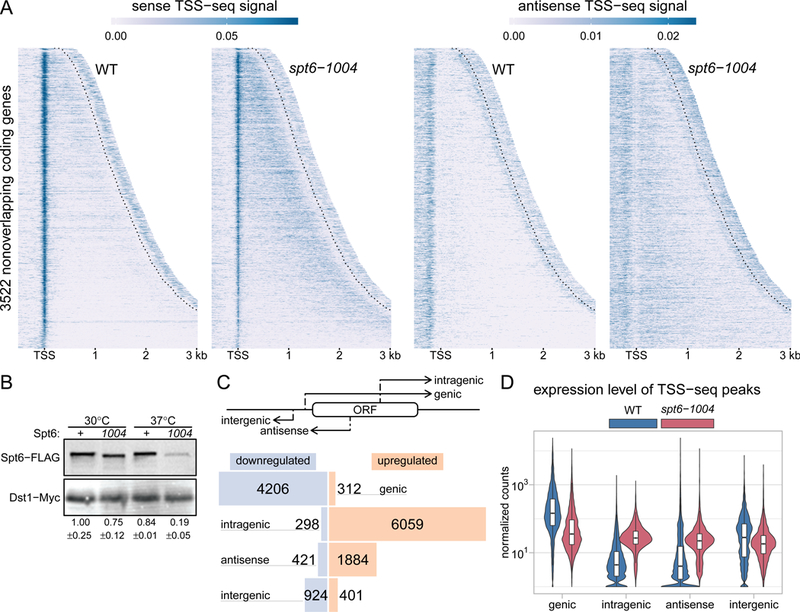
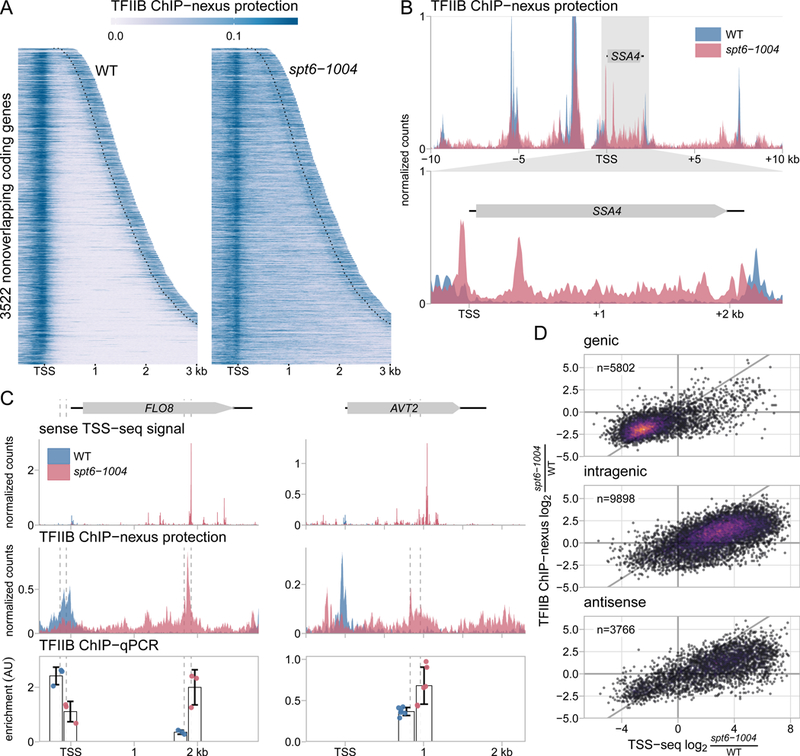
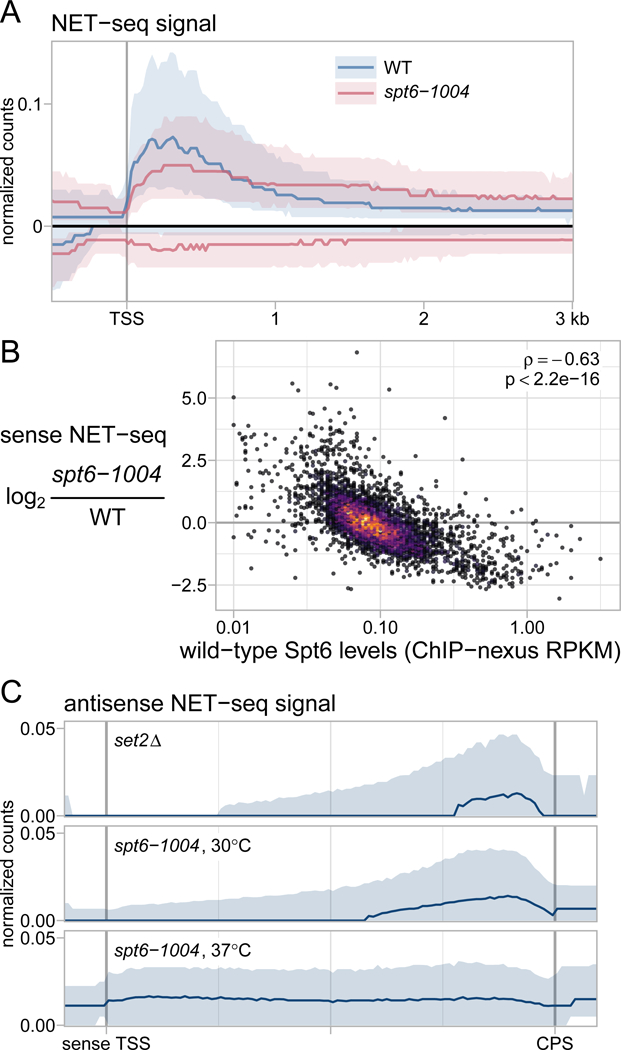
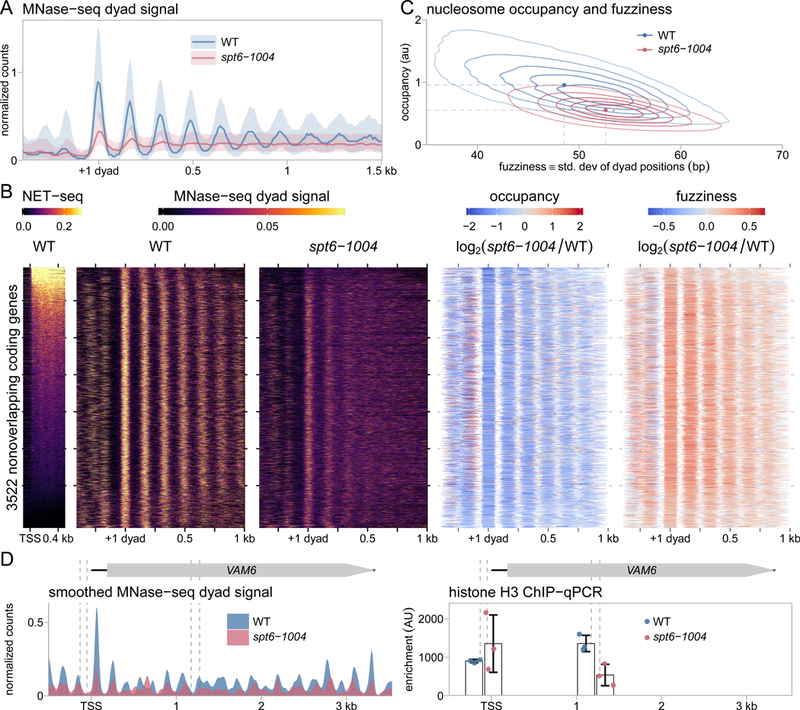
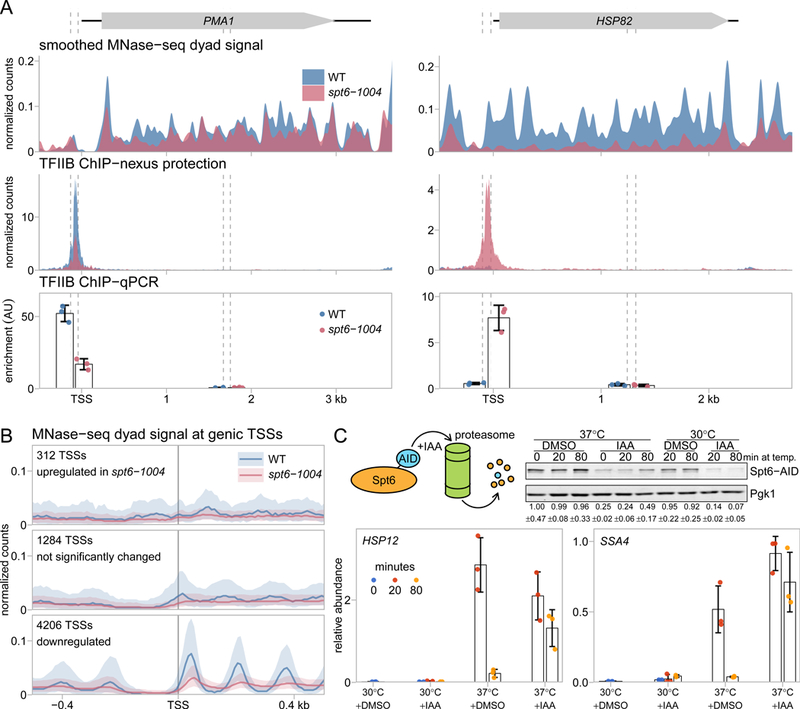
Spt6 is a conserved factor that controls transcription and chromatin structure across the genome. Although Spt6 is viewed as an elongation factor, spt6 mutations in Saccharomyces cerevisiae allow elevated levels of transcripts from within coding regions, suggesting that Spt6 also controls initiation. To address the requirements for Spt6 in transcription and chromatin structure, we have combined four genome-wide approaches. Our results demonstrate that Spt6 represses transcription initiation at thousands of intragenic promoters. We characterize these intragenic promoters and find sequence features conserved with genic promoters. Finally, we show that Spt6 also regulates transcription initiation at most genic promoters and propose a model of initiation site competition to account for this. Together, our results demonstrate that Spt6 controls the fidelity of transcription initiation throughout the genome.
Keywords: Spt6; chromatin structure; intragenic promoters; transcription start sites.
Copyright © 2018 Elsevier Inc. All rights reserved.
Conflict of interest statement
DECLARATION OF INTERESTS
The authors declare no competing interests.
Figures

References
-
- Adkins MW, and Tyler JK (2006). Transcriptional activators are dispensable for transcription in the absence of Spt6-mediated chromatin reassembly of promoter regions. Mol Cell 21, 405–416. - PubMed
-
- Andrews S (2014). FastQC: a quality control tool for high throughput sequence data. In Babraham Bioinformatics
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
