

High-quality genome sequences of uncultured microbes by assembly of read clouds
- PMID: 30320765
- PMCID: PMC6465186
- DOI: 10.1038/nbt.4266
High-quality genome sequences of uncultured microbes by assembly of read clouds
Abstract
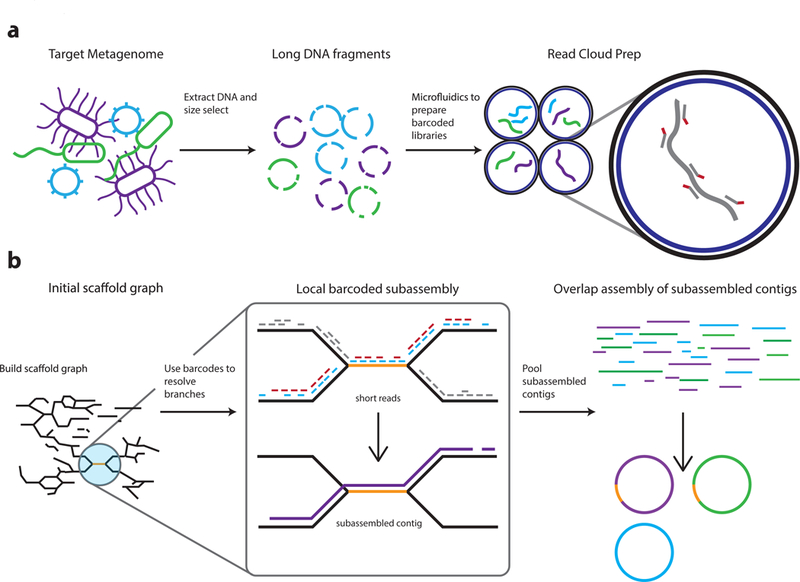
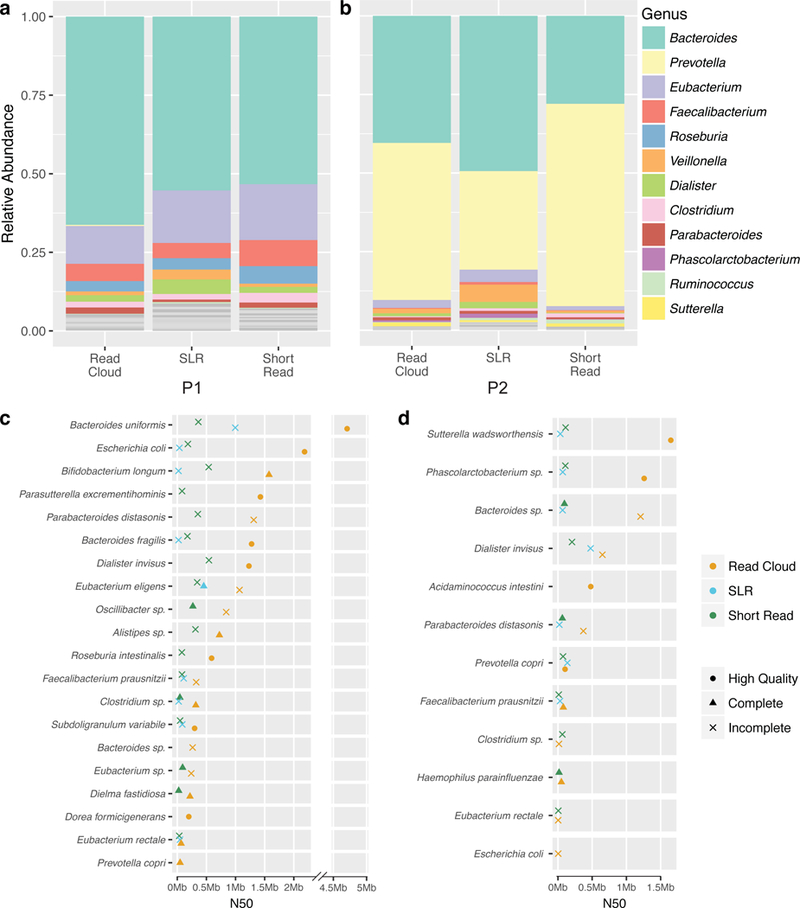
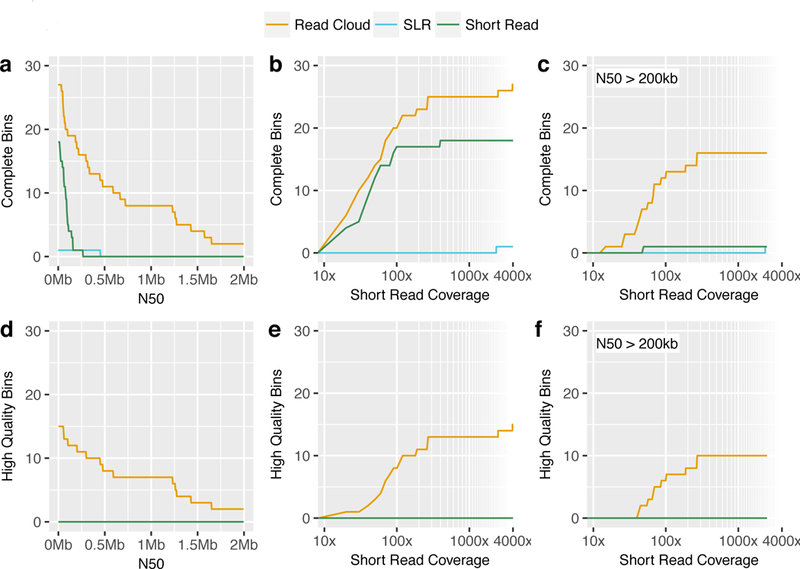
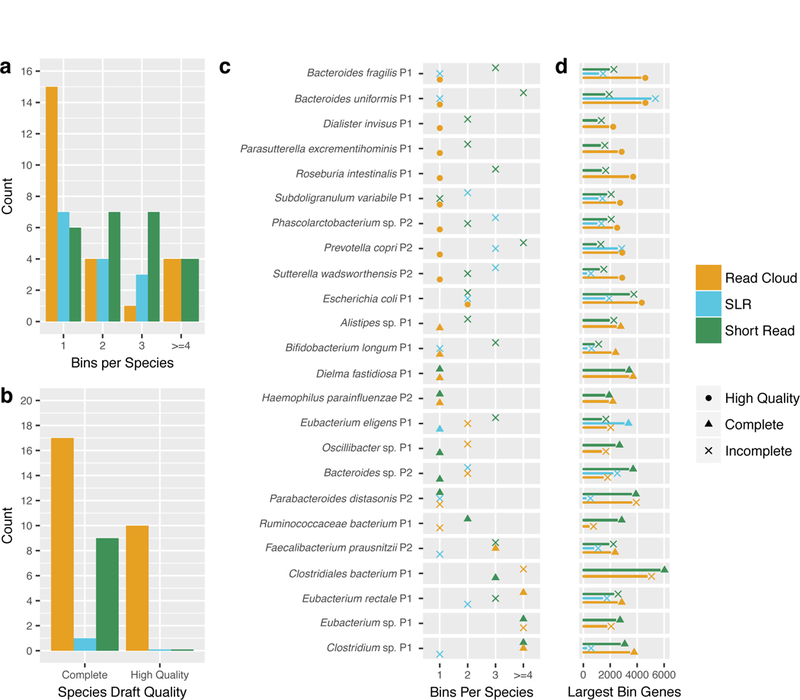
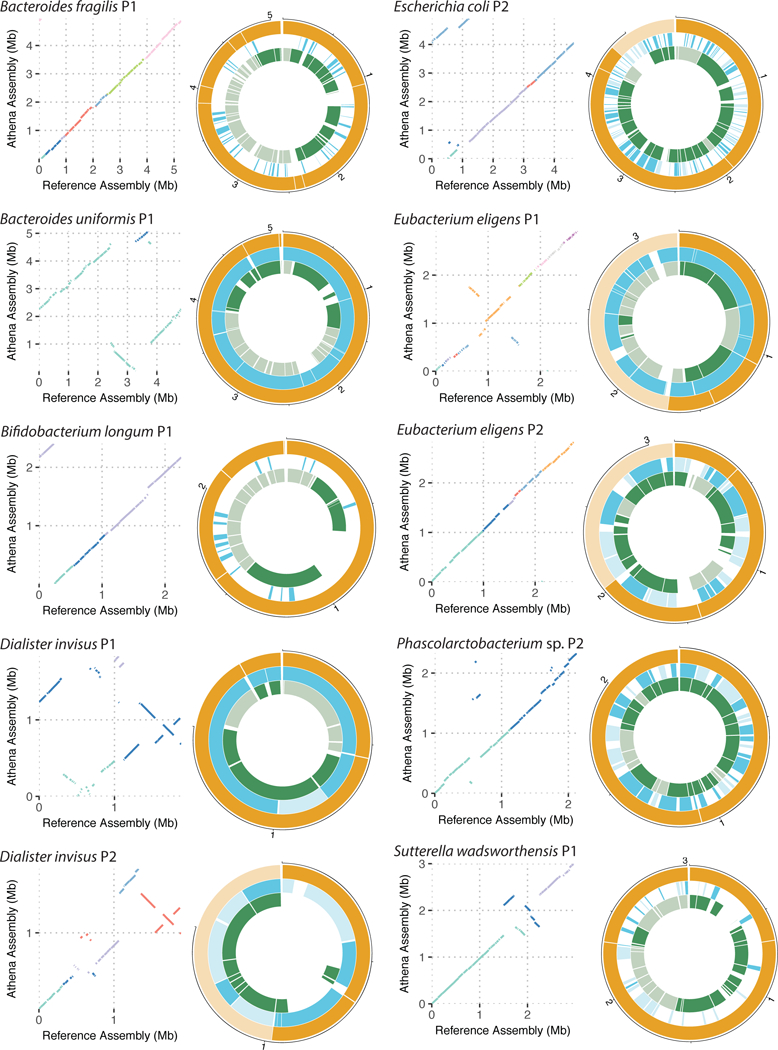
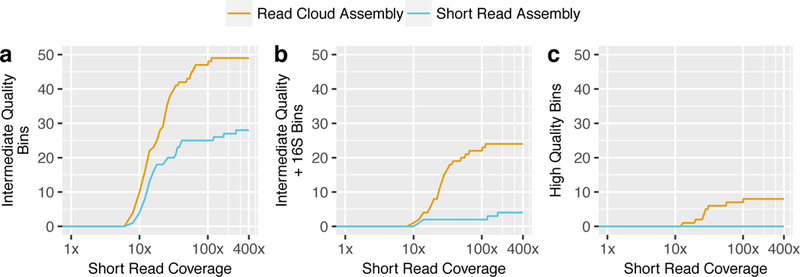
Although shotgun metagenomic sequencing of microbiome samples enables partial reconstruction of strain-level community structure, obtaining high-quality microbial genome drafts without isolation and culture remains difficult. Here, we present an application of read clouds, short-read sequences tagged with long-range information, to microbiome samples. We present Athena, a de novo assembler that uses read clouds to improve metagenomic assemblies. We applied this approach to sequence stool samples from two healthy individuals and compared it with existing short-read and synthetic long-read metagenomic sequencing techniques. Read-cloud metagenomic sequencing and Athena assembly produced the most comprehensive individual genome drafts with high contiguity (>200-kb N50, fewer than ten contigs), even for bacteria with relatively low (20×) raw short-read-sequence coverage. We also sequenced a complex marine-sediment sample and generated 24 intermediate-quality genome drafts (>70% complete, <10% contaminated), nine of which were complete (>90% complete, <5% contaminated). Our approach allows for culture-free generation of high-quality microbial genome drafts by using a single shotgun experiment.
Conflict of interest statement
Competing financial interests
S.B. is an employee and owns stock in Illumina. Shotgun sequencing products developed, marketed and/or sold by Illumina were used in this manuscript.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
