

Integrative Molecular Characterization of Malignant Pleural Mesothelioma
- PMID: 30322867
- PMCID: PMC6310008
- DOI: 10.1158/2159-8290.CD-18-0804
Integrative Molecular Characterization of Malignant Pleural Mesothelioma
Abstract
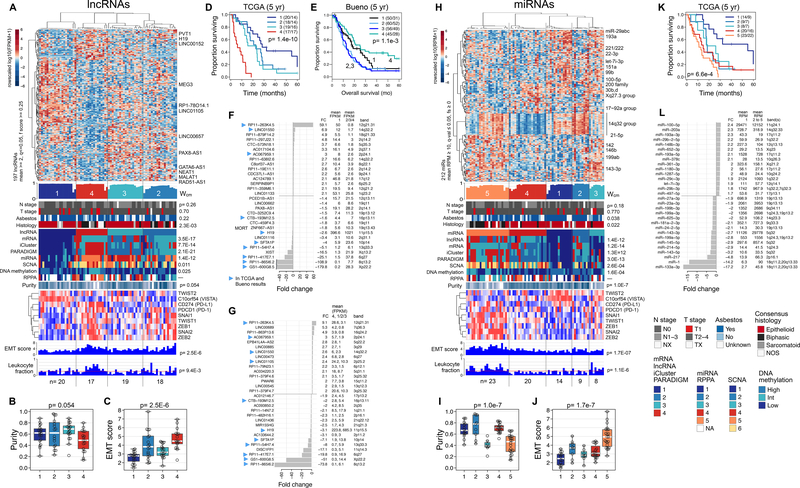
Malignant pleural mesothelioma (MPM) is a highly lethal cancer of the lining of the chest cavity. To expand our understanding of MPM, we conducted a comprehensive integrated genomic study, including the most detailed analysis of BAP1 alterations to date. We identified histology-independent molecular prognostic subsets, and defined a novel genomic subtype with TP53 and SETDB1 mutations and extensive loss of heterozygosity. We also report strong expression of the immune-checkpoint gene VISTA in epithelioid MPM, strikingly higher than in other solid cancers, with implications for the immune response to MPM and for its immunotherapy. Our findings highlight new avenues for further investigation of MPM biology and novel therapeutic options. SIGNIFICANCE: Through a comprehensive integrated genomic study of 74 MPMs, we provide a deeper understanding of histology-independent determinants of aggressive behavior, define a novel genomic subtype with TP53 and SETDB1 mutations and extensive loss of heterozygosity, and discovered strong expression of the immune-checkpoint gene VISTA in epithelioid MPM.See related commentary by Aggarwal and Albelda, p. 1508.This article is highlighted in the In This Issue feature, p. 1494.
©2018 American Association for Cancer Research.
Conflict of interest statement
Conflict of Interest
Andrew Cherniak and Matthew Meyerson received funding from Bayer AG. Ewan Gibb is an employee of GenomeDX Biosciences, INC. Harvey Pass’ spouse is on the Speakers Board for Genentec, and Genentec has performed molecular analyses of a separate MPM cohort for Dr. Pass’ work.
Figures






Comment in
-
Molecular Characterization of Malignant Mesothelioma: Time for New Targets?Cancer Discov. 2018 Dec;8(12):1508-1510. doi: 10.1158/2159-8290.CD-18-1181. Cancer Discov. 2018. PMID: 30510013
-
Mesothelioma in the age of "Omics": Before and after The Cancer Genome Atlas.J Thorac Cardiovasc Surg. 2020 Oct;160(4):1078-1083.e2. doi: 10.1016/j.jtcvs.2020.02.141. Epub 2020 May 29. J Thorac Cardiovasc Surg. 2020. PMID: 32475501 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
- P30 CA016672/CA/NCI NIH HHS/United States
- U24 CA143882/CA/NCI NIH HHS/United States
- U24 CA143866/CA/NCI NIH HHS/United States
- U54 HG003273/HG/NHGRI NIH HHS/United States
- U24 CA143858/CA/NCI NIH HHS/United States
- U24 CA143848/CA/NCI NIH HHS/United States
- U54 HG003079/HG/NHGRI NIH HHS/United States
- R00 CA207871/CA/NCI NIH HHS/United States
- T32 CA009666/CA/NCI NIH HHS/United States
- U24 CA210990/CA/NCI NIH HHS/United States
- U54 HG003067/HG/NHGRI NIH HHS/United States
- U24 CA143835/CA/NCI NIH HHS/United States
- U24 CA210950/CA/NCI NIH HHS/United States
- U24 CA143845/CA/NCI NIH HHS/United States
- U24 CA143799/CA/NCI NIH HHS/United States
- P30 CA008748/CA/NCI NIH HHS/United States
- T32 HG008345/HG/NHGRI NIH HHS/United States
- U24 CA144025/CA/NCI NIH HHS/United States
- U24 CA143840/CA/NCI NIH HHS/United States
- R01 CA120528/CA/NCI NIH HHS/United States
- U24 CA143843/CA/NCI NIH HHS/United States
- K99 CA207871/CA/NCI NIH HHS/United States
- U24 CA210969/CA/NCI NIH HHS/United States
- U24 CA143883/CA/NCI NIH HHS/United States
- U24 CA143867/CA/NCI NIH HHS/United States
- U24 CA199461/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
