

ADP-ribosyl-binding and hydrolase activities of the alphavirus nsP3 macrodomain are critical for initiation of virus replication
- PMID: 30322911
- PMCID: PMC6217424
- DOI: 10.1073/pnas.1812130115
ADP-ribosyl-binding and hydrolase activities of the alphavirus nsP3 macrodomain are critical for initiation of virus replication
Abstract
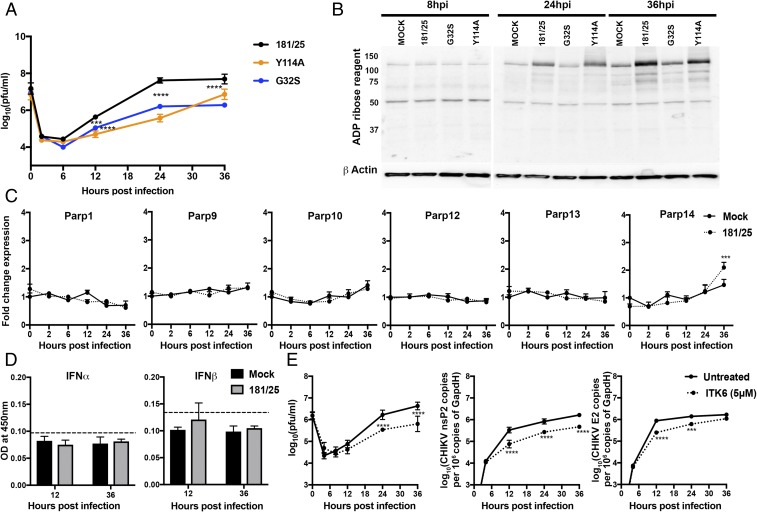
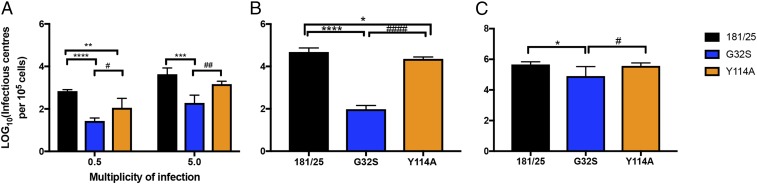
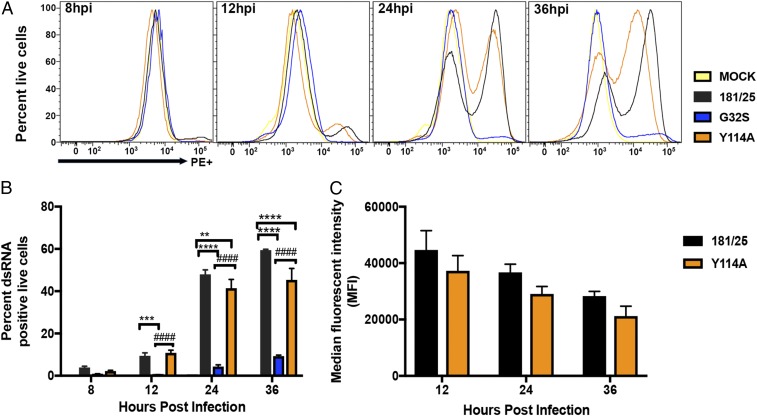
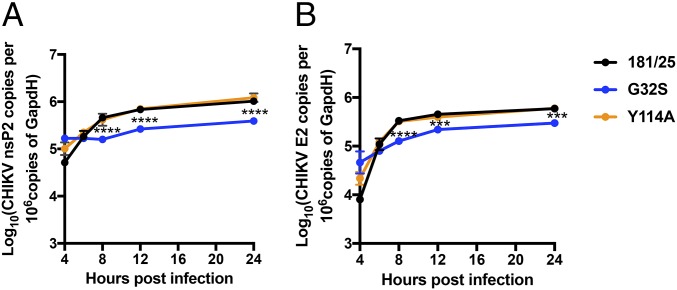
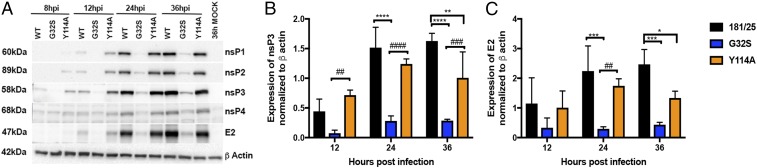
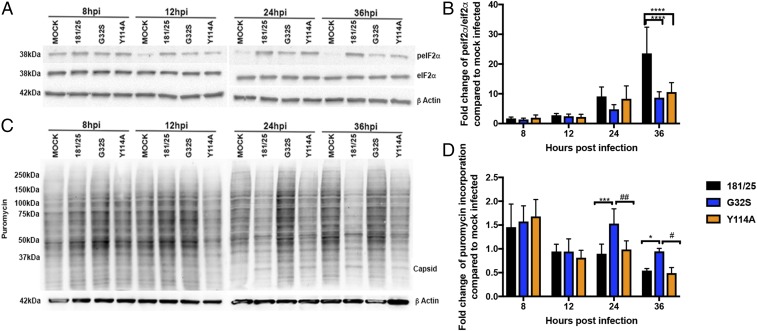
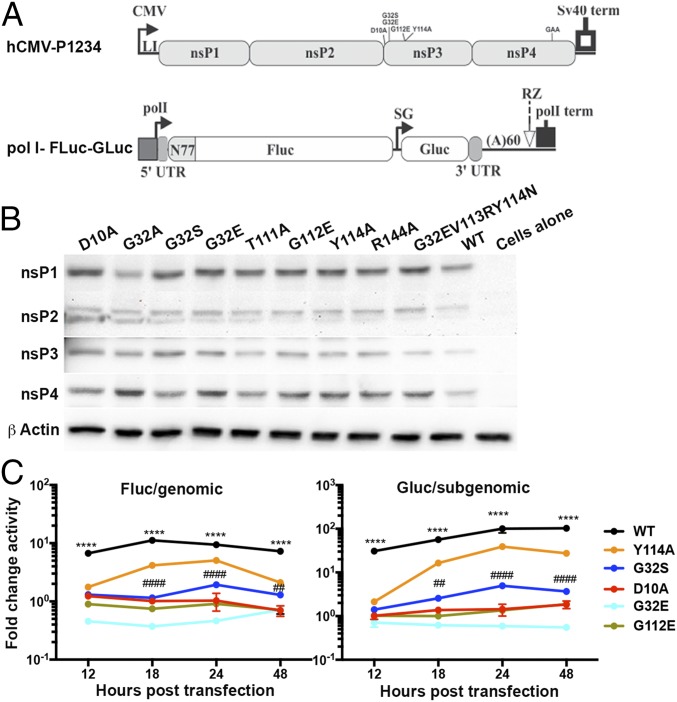
Alphaviruses are plus-strand RNA viruses that cause encephalitis, rash, and arthritis. The nonstructural protein (nsP) precursor polyprotein is translated from genomic RNA and processed into four nsPs. nsP3 has a highly conserved macrodomain (MD) that binds ADP-ribose (ADPr), which can be conjugated to protein as a posttranslational modification involving transfer of ADPr from NAD+ by poly ADPr polymerases (PARPs). The nsP3MD also removes ADPr from mono ADP-ribosylated (MARylated) substrates. To determine which aspects of alphavirus replication require nsP3MD ADPr-binding and/or hydrolysis function, we studied NSC34 neuronal cells infected with chikungunya virus (CHIKV). Infection induced ADP-ribosylation of cellular proteins without increasing PARP expression, and inhibition of MARylation decreased virus replication. CHIKV with a G32S mutation that reduced ADPr-binding and hydrolase activities was less efficient than WT CHIKV in establishing infection and in producing nsPs, dsRNA, viral RNA, and infectious virus. CHIKV with a Y114A mutation that increased ADPr binding but reduced hydrolase activity, established infection like WT CHIKV, rapidly induced nsP translation, and shut off host protein synthesis with reduced amplification of dsRNA. To assess replicase function independent of virus infection, a transreplicase system was used. Mutant nsP3MDs D10A, G32E, and G112E with no binding or hydrolase activity had no replicase activity, G32S had little, and Y114A was intermediate to WT. Therefore, ADP ribosylation of proteins and nsP3MD ADPr binding are necessary for initiation of alphavirus replication, while hydrolase activity facilitates amplification of replication complexes. These observations are consistent with observed nsP3MD conservation and limited tolerance for mutation.
Keywords: ADP ribosylation; PARP; alphavirus; macrodomain; replication complexes.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Gubler DJ. The global emergence/resurgence of arboviral diseases as public health problems. Arch Med Res. 2002;33:330–342. - PubMed
-
- Weaver SC, Lecuit M. Chikungunya virus and the global spread of a mosquito-borne disease. N Engl J Med. 2015;372:1231–1239. - PubMed
-
- Chandak NH, et al. Neurological complications of chikungunya virus infection. Neurol India. 2009;57:177–180. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
