

Firefly genomes illuminate parallel origins of bioluminescence in beetles
- PMID: 30324905
- PMCID: PMC6191289
- DOI: 10.7554/eLife.36495
Firefly genomes illuminate parallel origins of bioluminescence in beetles
Abstract
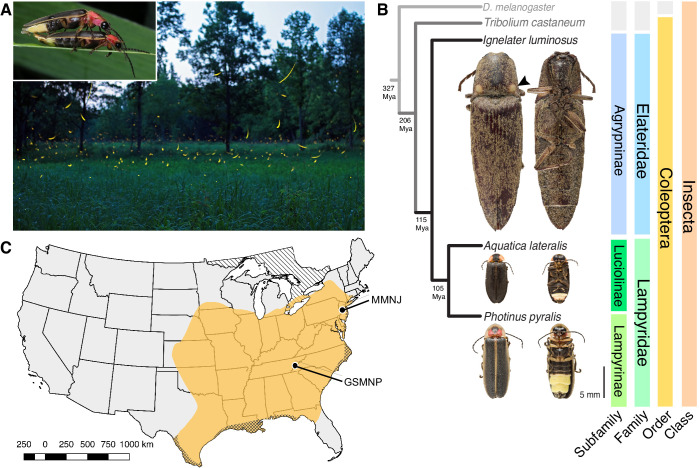
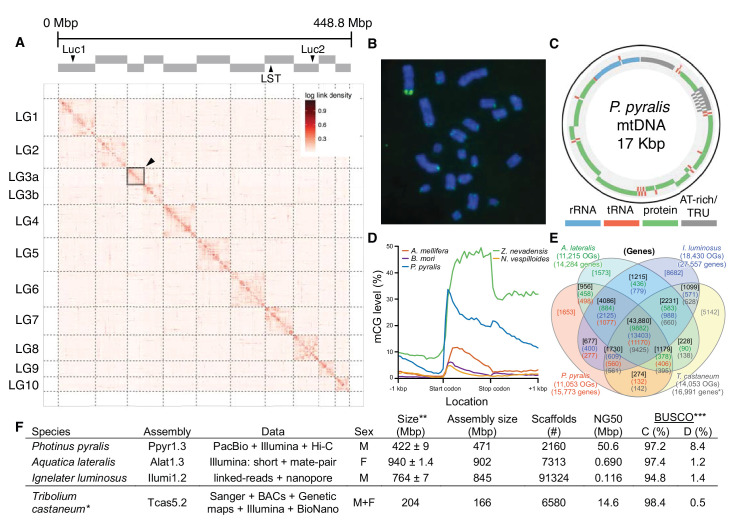
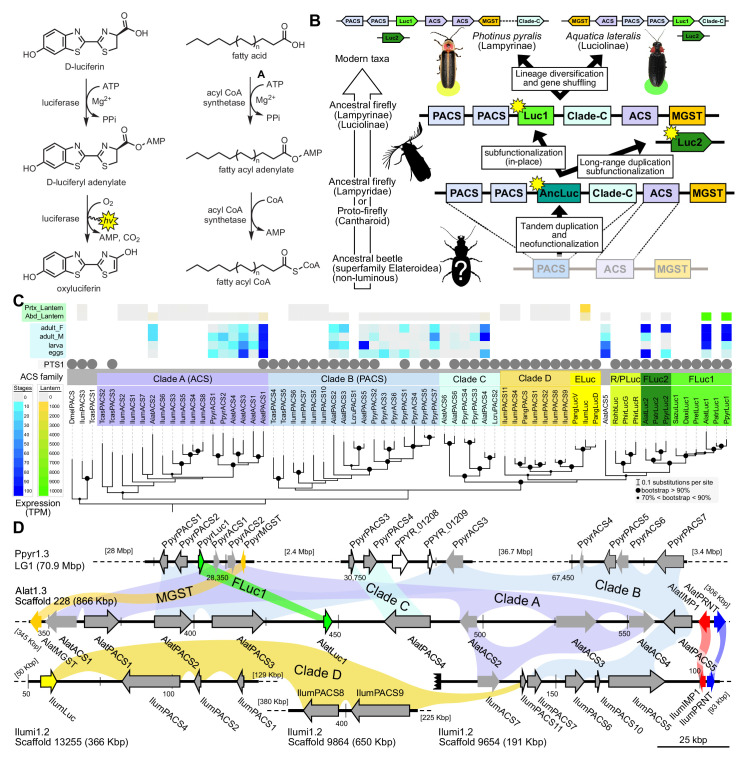
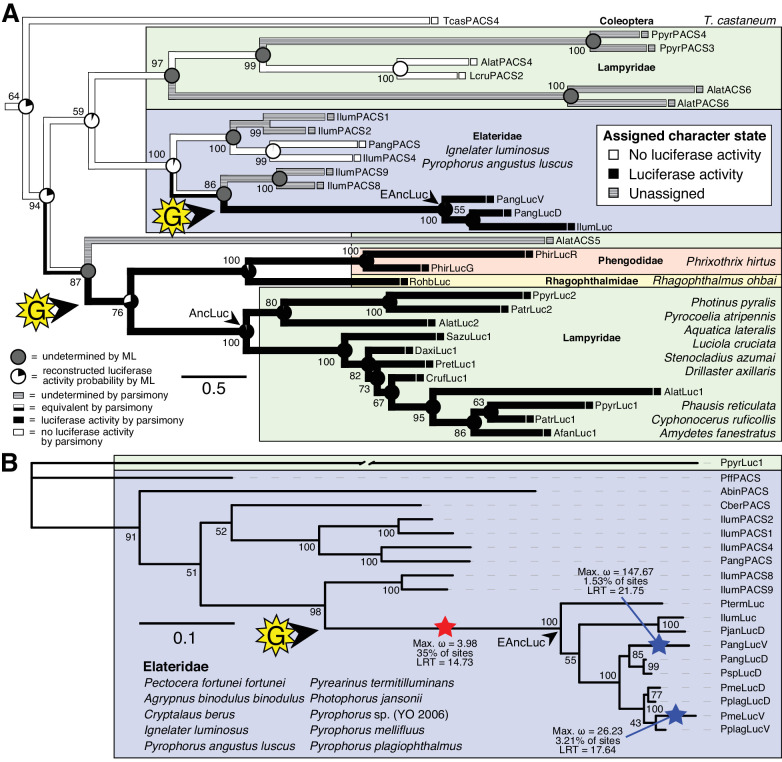
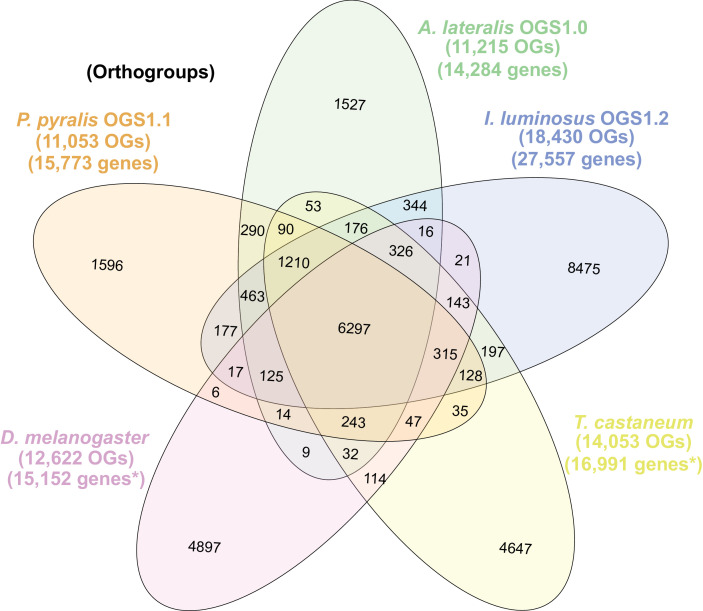
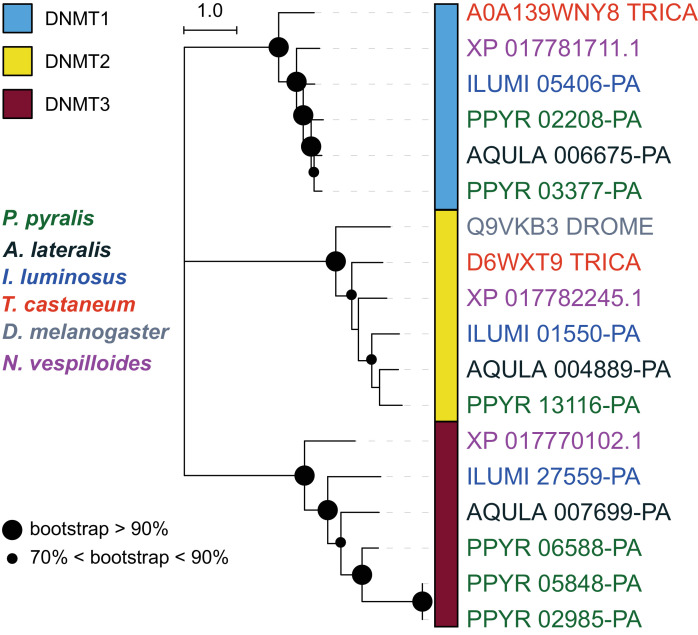
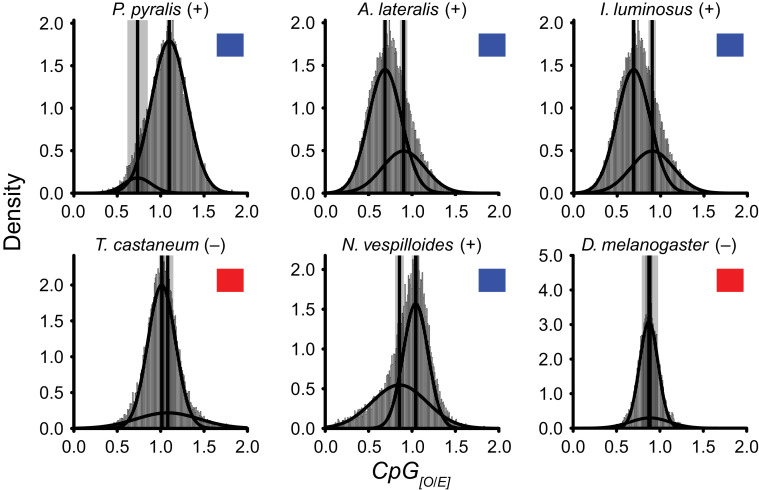
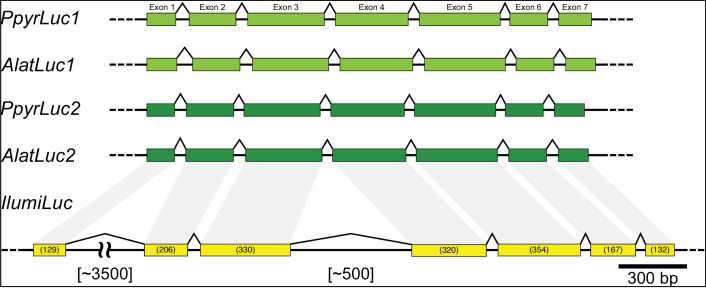
Fireflies and their luminous courtships have inspired centuries of scientific study. Today firefly luciferase is widely used in biotechnology, but the evolutionary origin of bioluminescence within beetles remains unclear. To shed light on this long-standing question, we sequenced the genomes of two firefly species that diverged over 100 million-years-ago: the North American Photinus pyralis and Japanese Aquatica lateralis. To compare bioluminescent origins, we also sequenced the genome of a related click beetle, the Caribbean Ignelater luminosus, with bioluminescent biochemistry near-identical to fireflies, but anatomically unique light organs, suggesting the intriguing hypothesis of parallel gains of bioluminescence. Our analyses support independent gains of bioluminescence in fireflies and click beetles, and provide new insights into the genes, chemical defenses, and symbionts that evolved alongside their luminous lifestyle.
Keywords: Aquatica lateralis; Ignelater luminosus; Photinus pyralis; bioluminescence; firefly; genetics; genomics; luciferase.
© 2018, Fallon et al.
Conflict of interest statement
TF, SL, CC, MB, GM, AB, MB, HD, IW, JD, AS, CS, KS, DH, RS, DN, SL, SS, SB, AL, YO, JW No competing interests declared
Figures
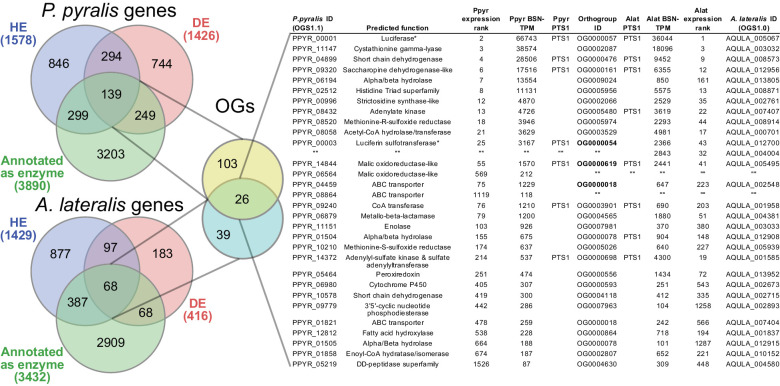
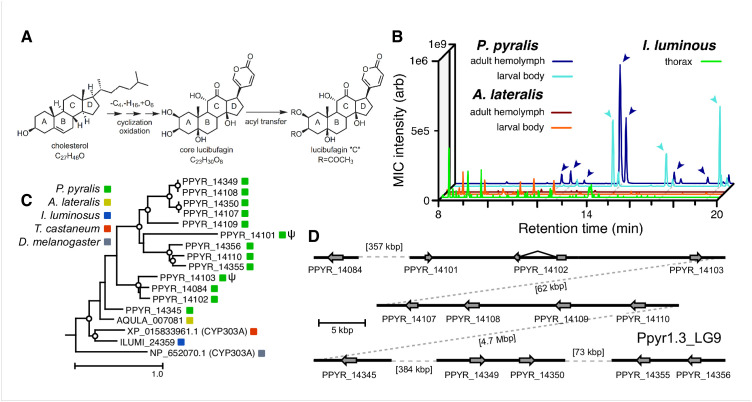
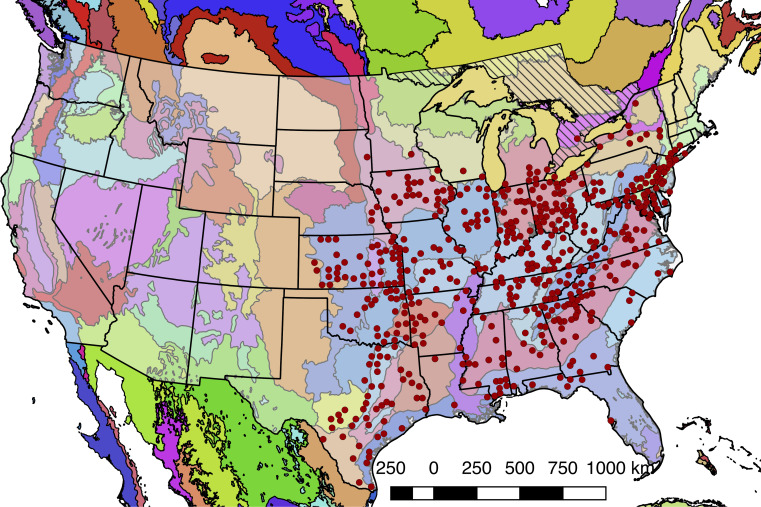




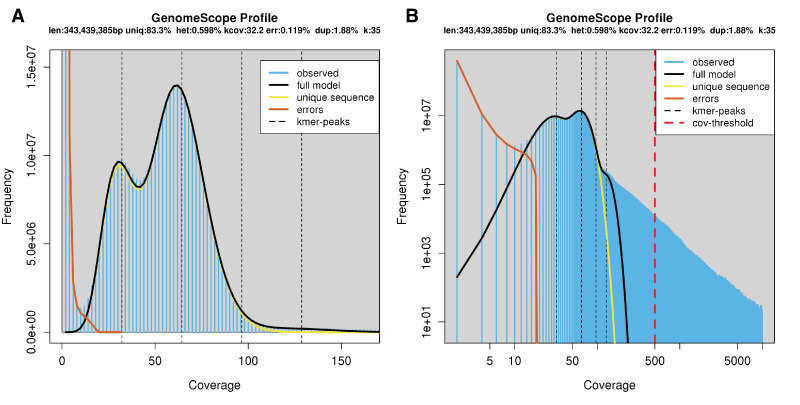
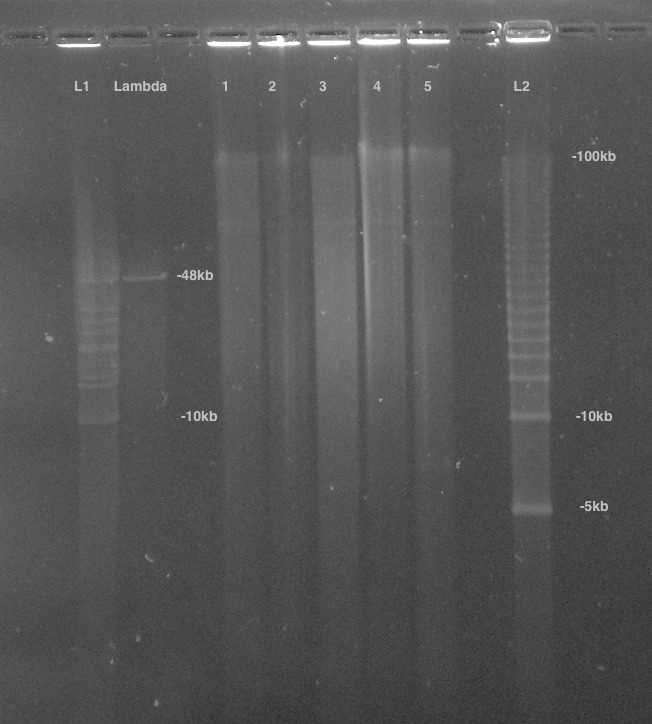

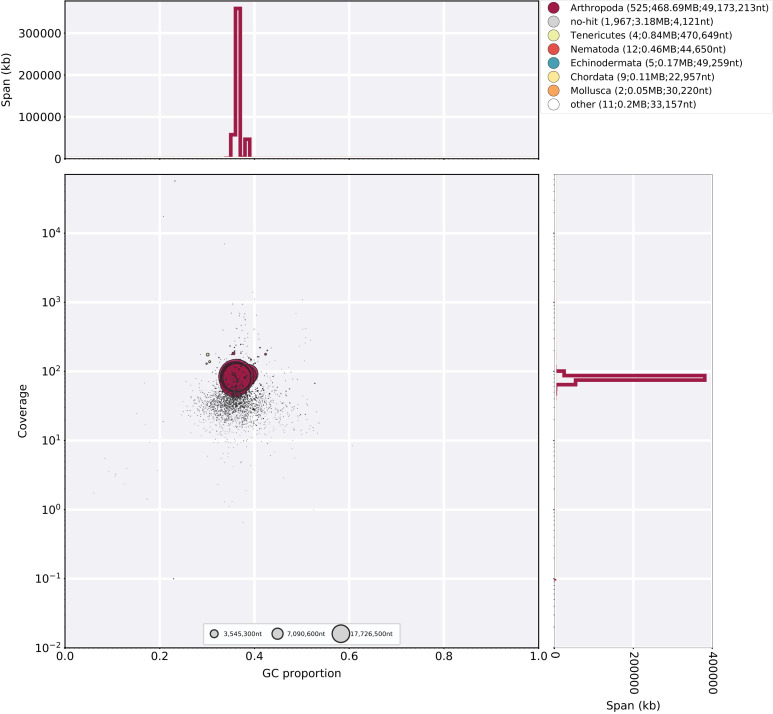
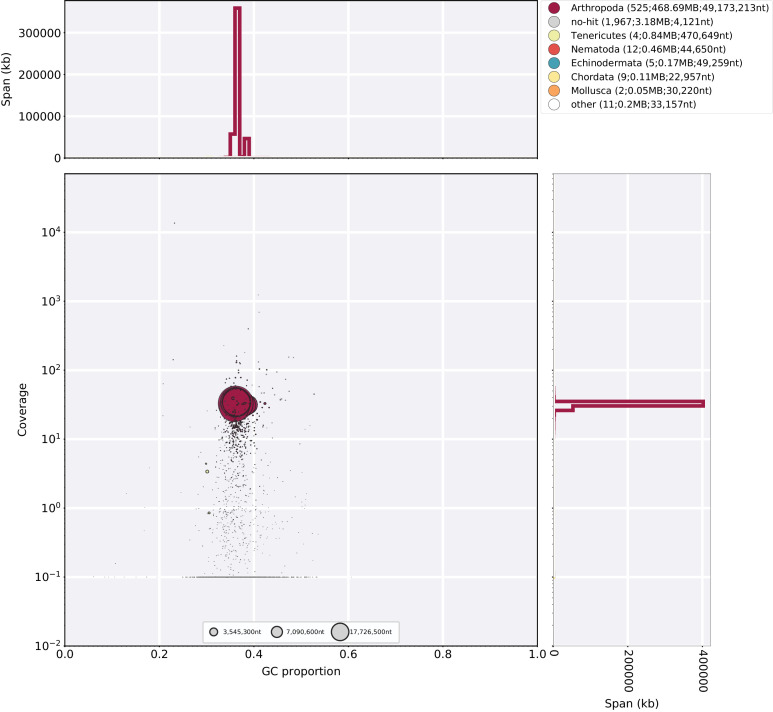
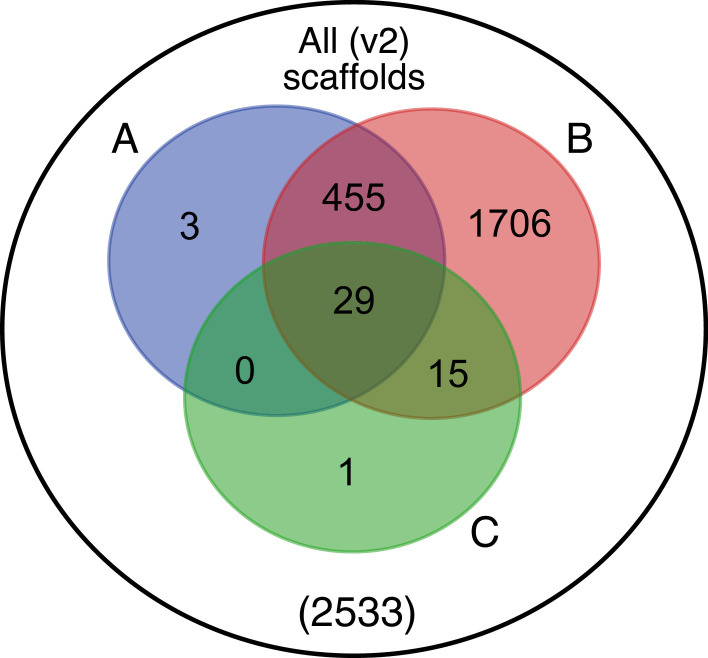

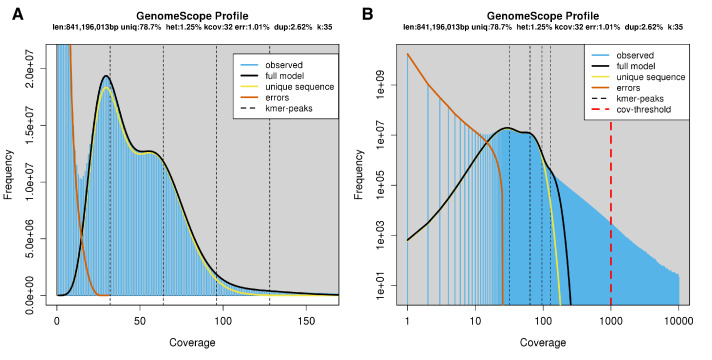
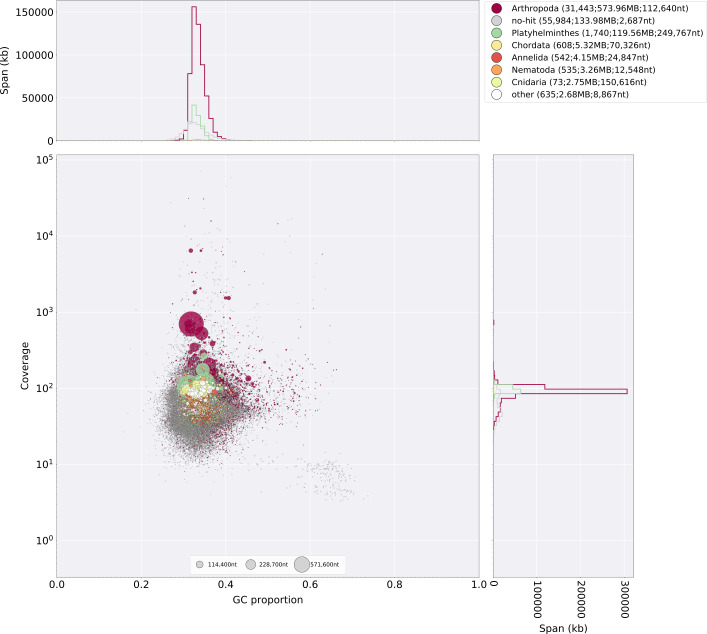

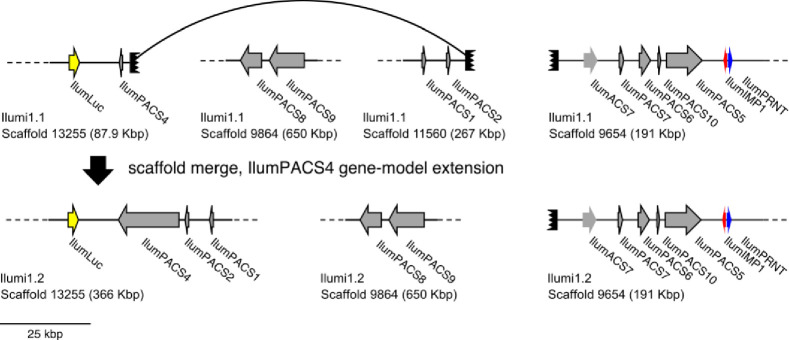
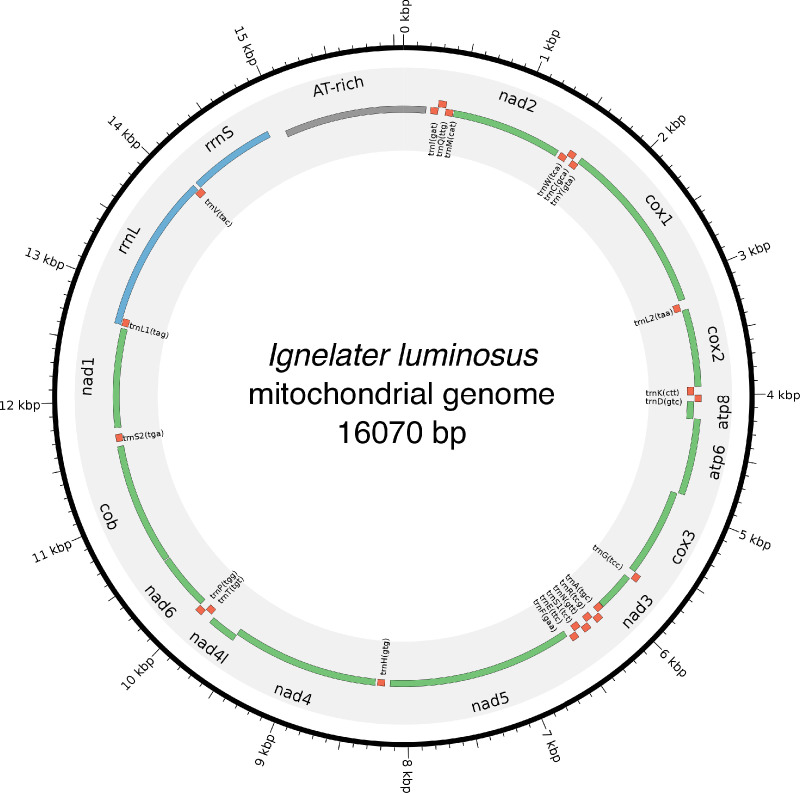
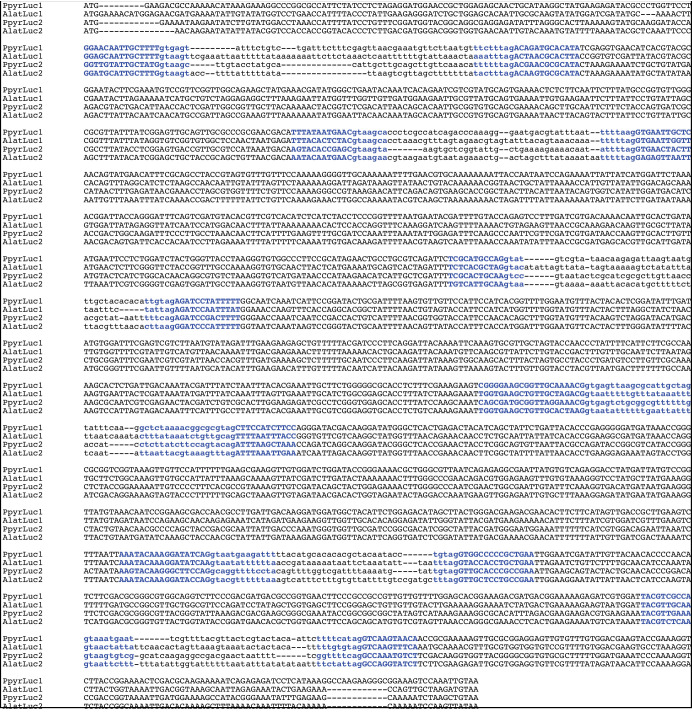
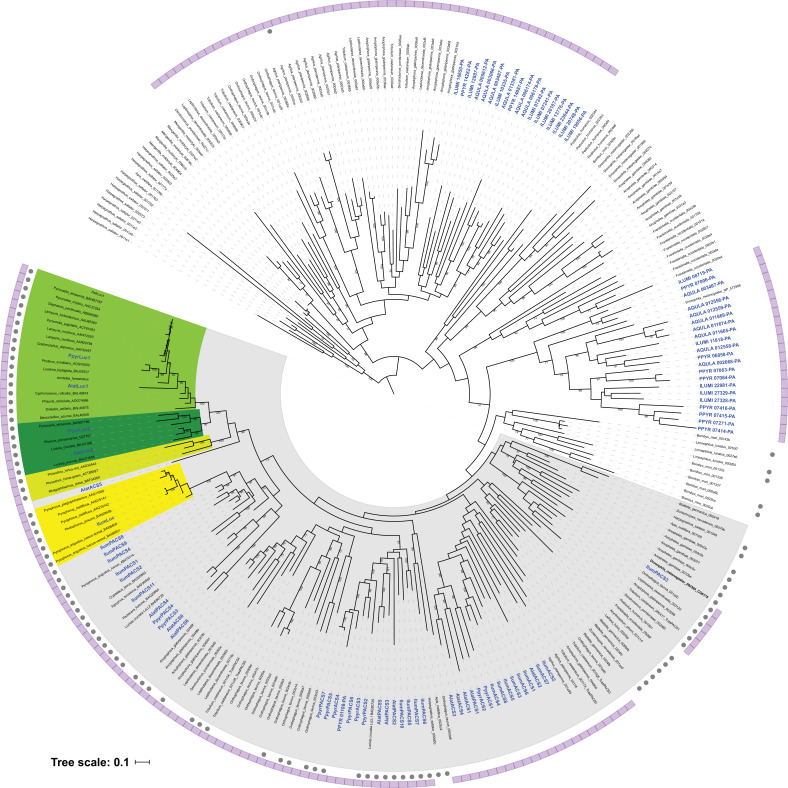

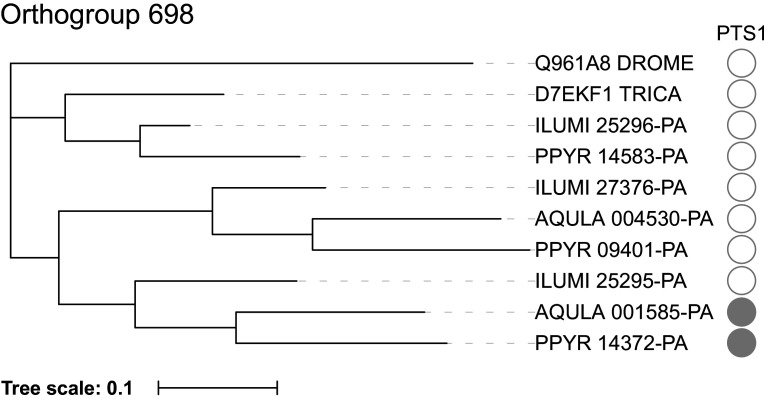
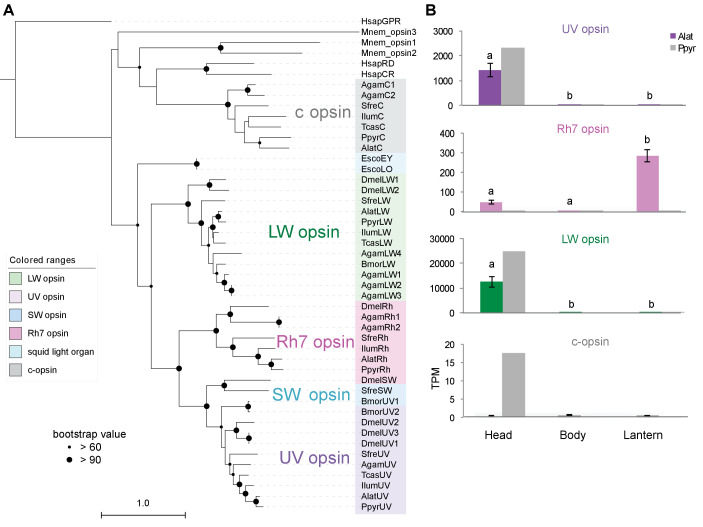
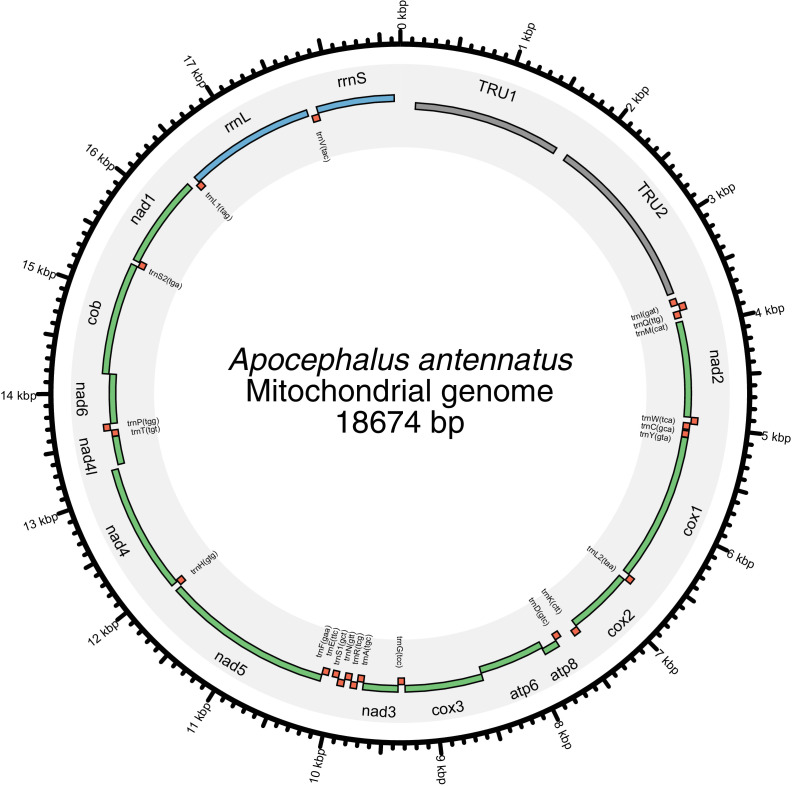
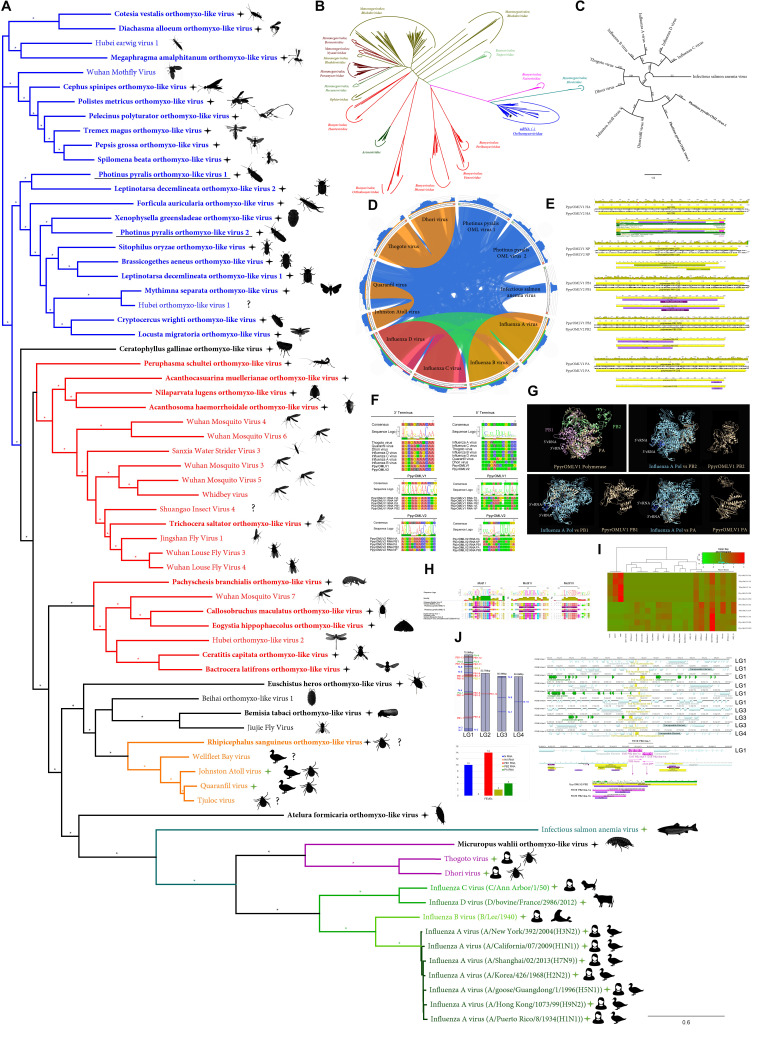
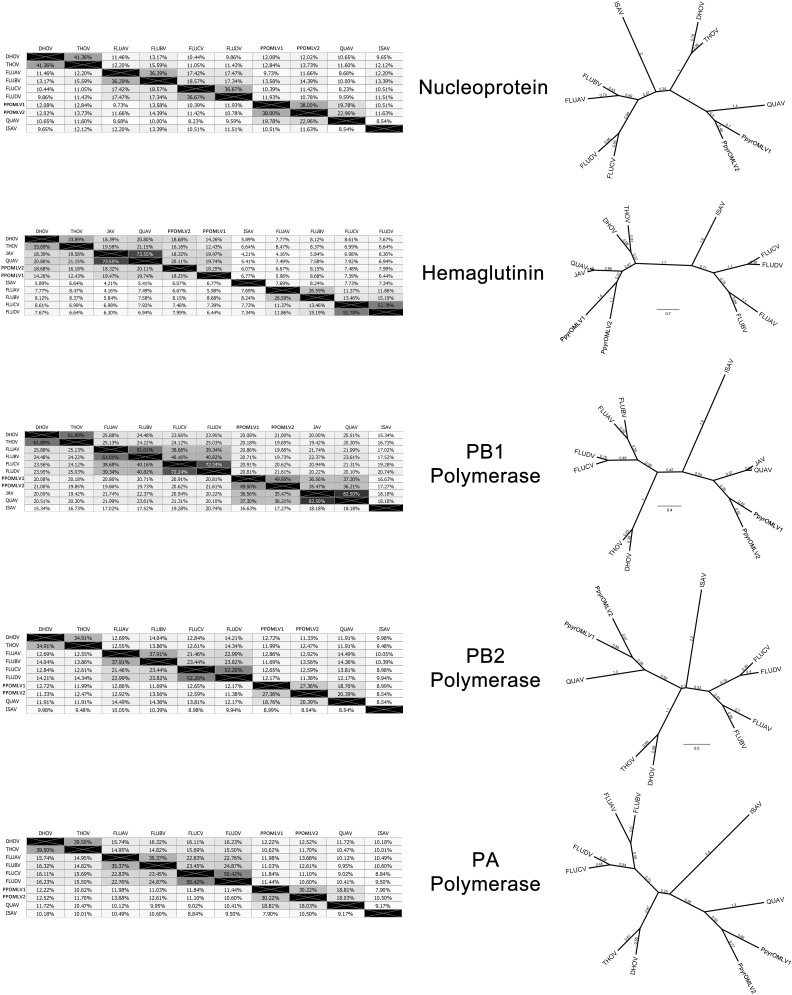
References
-
- Amaral DT, Silva JR, Viviani VR. Transcriptional comparison of the photogenic and non-photogenic tissues of Phrixothrix hirtus (Coleoptera: Phengodidae) and non-luminescent Chauliognathus flavipes (Coleoptera: Cantharidae) give insights on the origin of lanterns in railroad worms. Gene Reports. 2017;7:78–86. doi: 10.1016/j.genrep.2017.02.004. - DOI
-
- Anderson CR, Casals J. Dhori virus, a new agent isolated from Hyalomma dromedarii in India. The Indian Journal of Medical Research. 1973;61:1416–1420. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- T32 GM007287/GM/NIGMS NIH HHS/United States
- Pew Scholar Program in the Biomedical Sciences/Pew Charitable Trusts/International
- Lars G. Ljungdahl Distinguished Investigator/Georgia Research Alliance/International
- Graduate Student Fellowship/National Science Foundation/International
- https://experiment.com/projects/illuminating-the-firefly-genome/Experiment.com/International
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
