

Pathophysiology of Sickle Cell Disease
- PMID: 30332562
- PMCID: PMC7053558
- DOI: 10.1146/annurev-pathmechdis-012418-012838
Pathophysiology of Sickle Cell Disease
Abstract
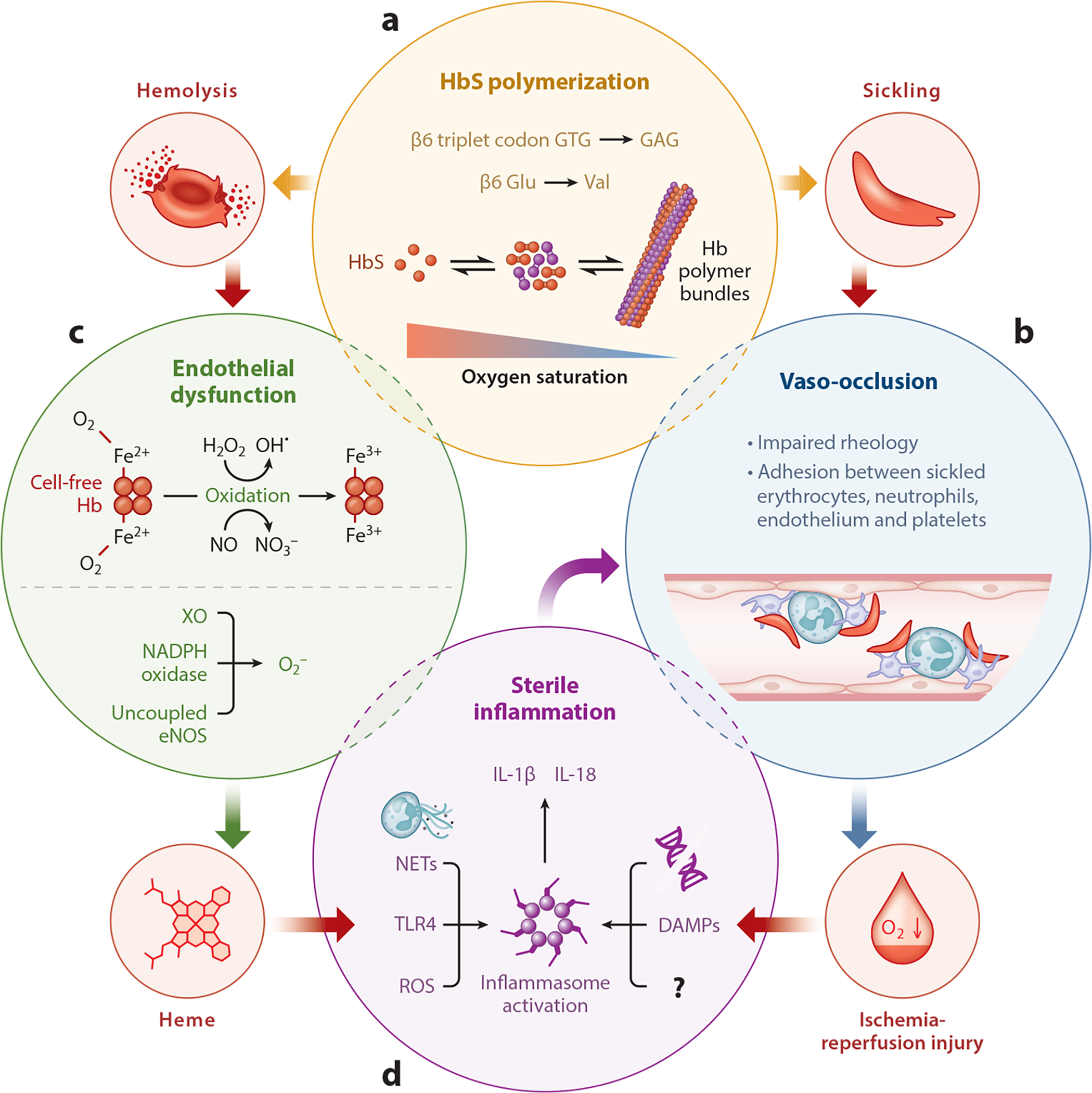
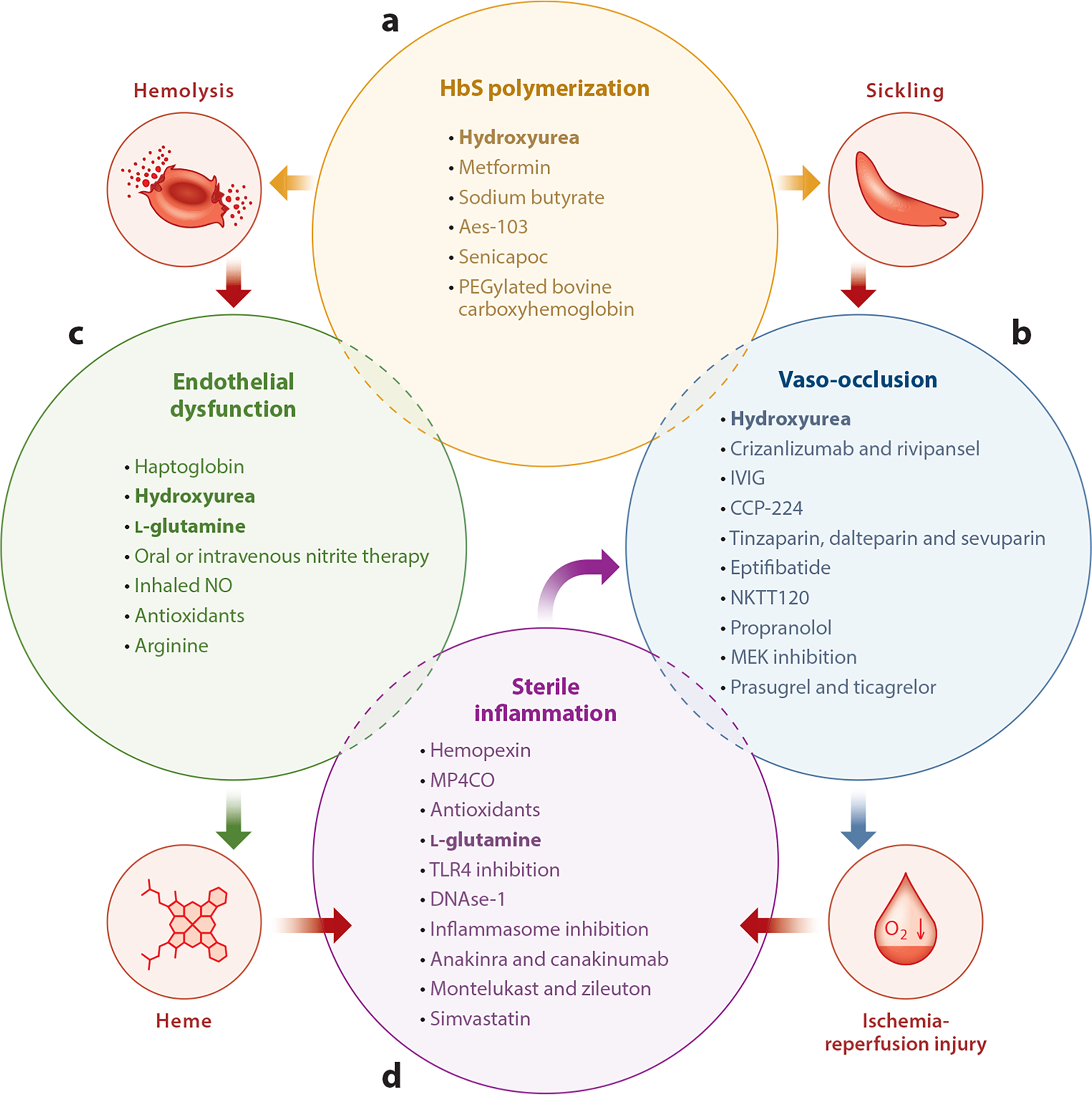
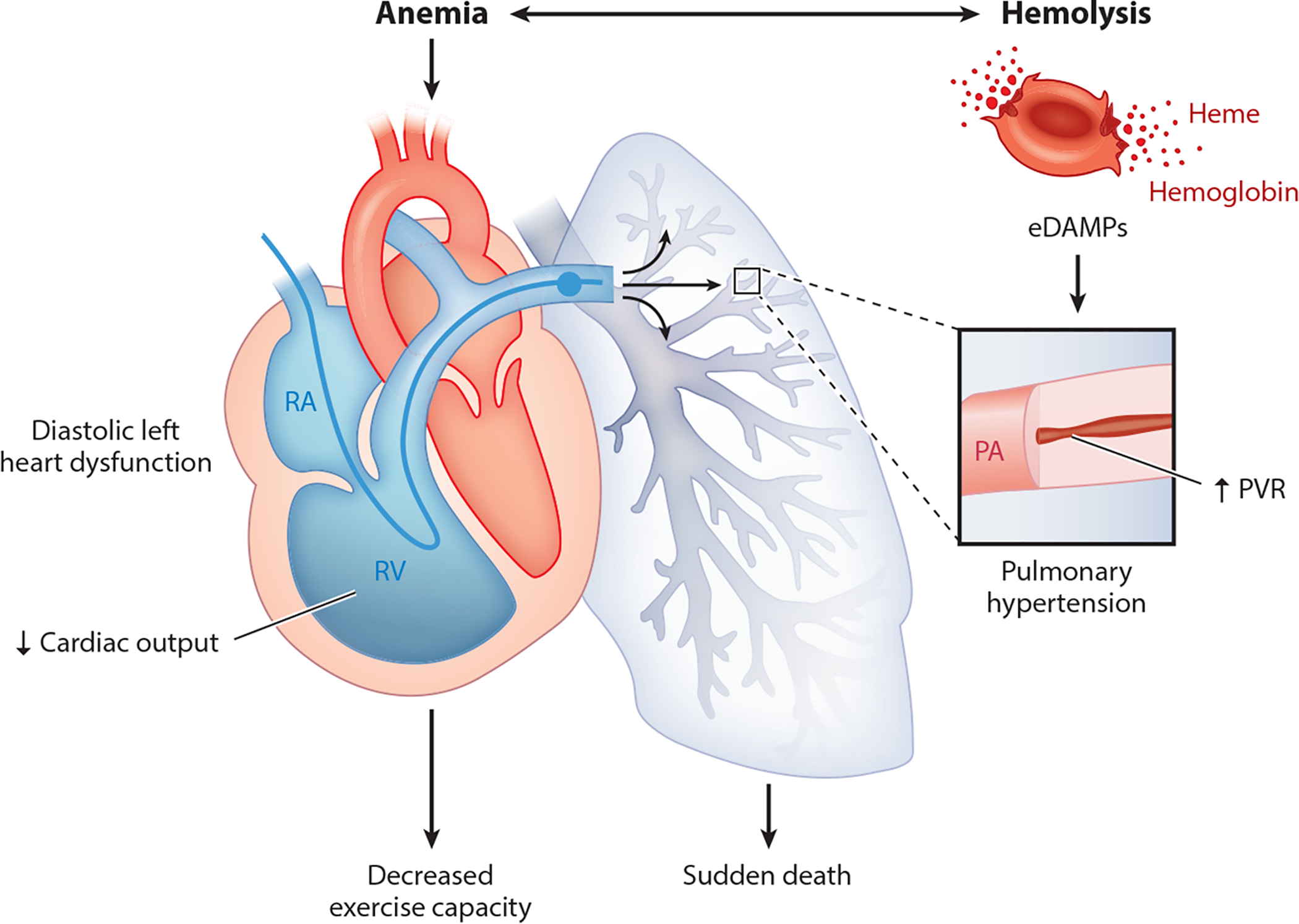
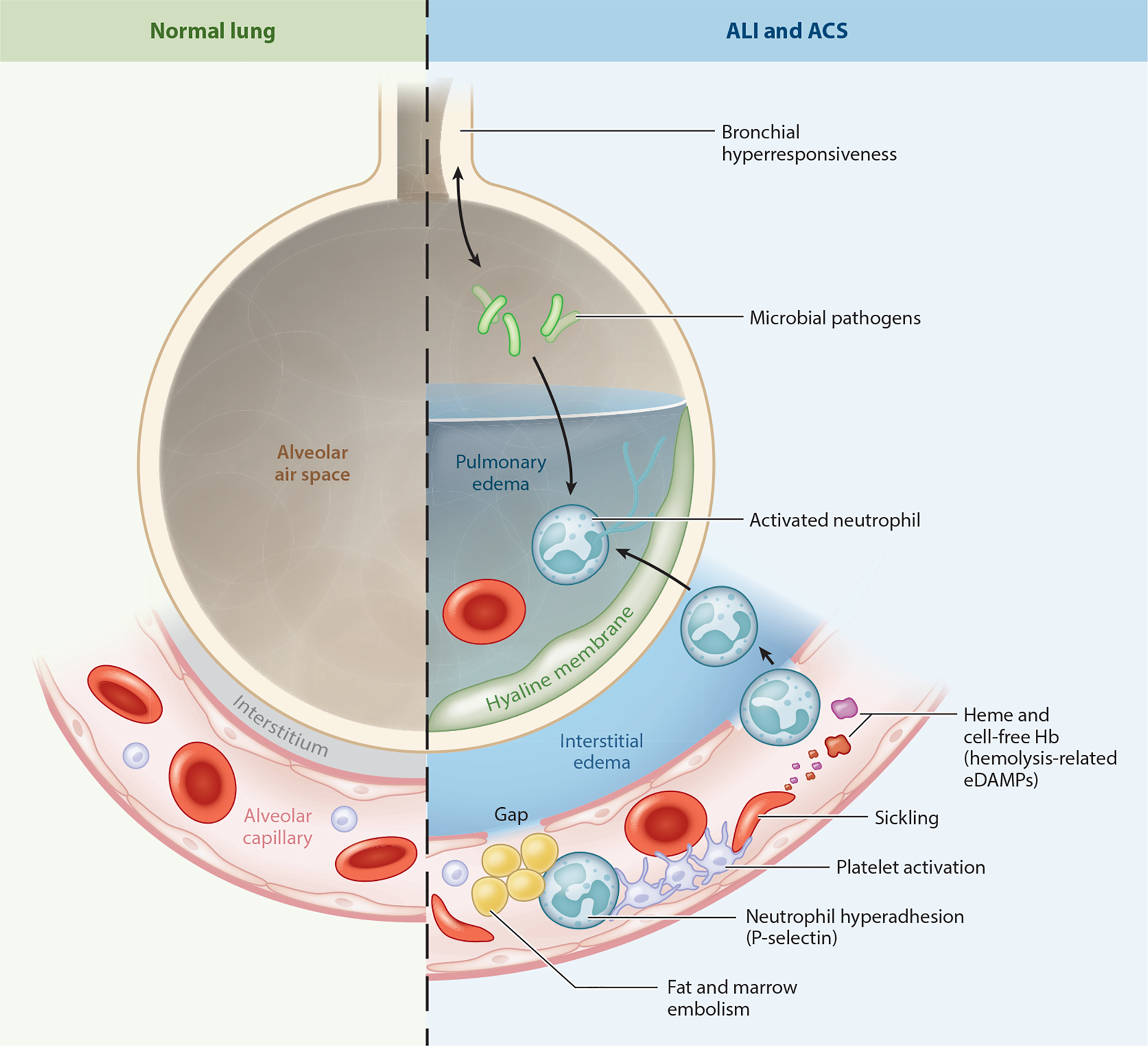
Since the discovery of sickle cell disease (SCD) in 1910, enormous strides have been made in the elucidation of the pathogenesis of its protean complications, which has inspired recent advances in targeted molecular therapies. In SCD, a single amino acid substitution in the β-globin chain leads to polymerization of mutant hemoglobin S, impairing erythrocyte rheology and survival. Clinically, erythrocyte abnormalities in SCD manifest in hemolytic anemia and cycles of microvascular vaso-occlusion leading to end-organ ischemia-reperfusion injury and infarction. Vaso-occlusive events and intravascular hemolysis promote inflammation and redox instability that lead to progressive small- and large-vessel vasculopathy. Based on current evidence, the pathobiology of SCD is considered to be a vicious cycle of four major processes, all the subject of active study and novel therapeutic targeting: ( a) hemoglobin S polymerization, ( b) impaired biorheology and increased adhesion-mediated vaso-occlusion, ( c) hemolysis-mediated endothelial dysfunction, and ( d) concerted activation of sterile inflammation (Toll-like receptor 4- and inflammasome-dependent innate immune pathways). These molecular, cellular, and biophysical processes synergize to promote acute and chronic pain and end-organ injury and failure in SCD. This review provides an exhaustive overview of the current understanding of the molecular pathophysiology of SCD, how this pathophysiology contributes to complications of the central nervous and cardiopulmonary systems, and how this knowledge is being harnessed to develop current and potential therapies.
Keywords: hemolysis; infarction; inflammation; oxidative stress; reperfusion injury; sickle cell anemia.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
