

A highly parallel strategy for storage of digital information in living cells
- PMID: 30333005
- PMCID: PMC6191901
- DOI: 10.1186/s12896-018-0476-4
A highly parallel strategy for storage of digital information in living cells
Abstract
Background: Encoding arbitrary digital information in DNA has attracted attention as a potential avenue for large scale and long term data storage. However, in order to enable DNA data storage technologies there needs to be improvements in data storage fidelity (tolerance to mutation), the facility of writing and reading the data (biases and systematic error arising from synthesis and sequencing), and overall scalability.
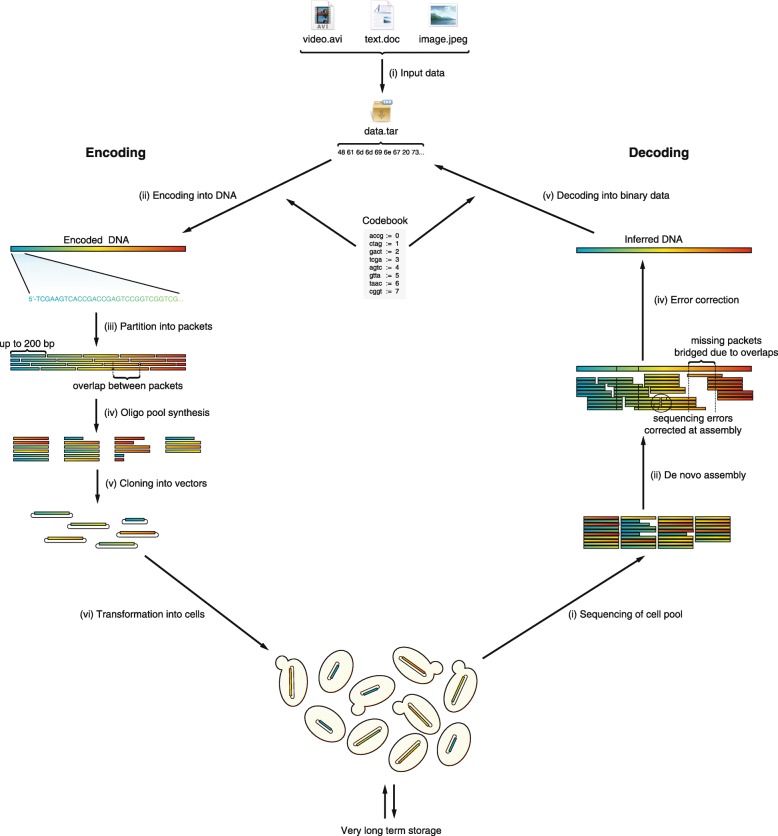
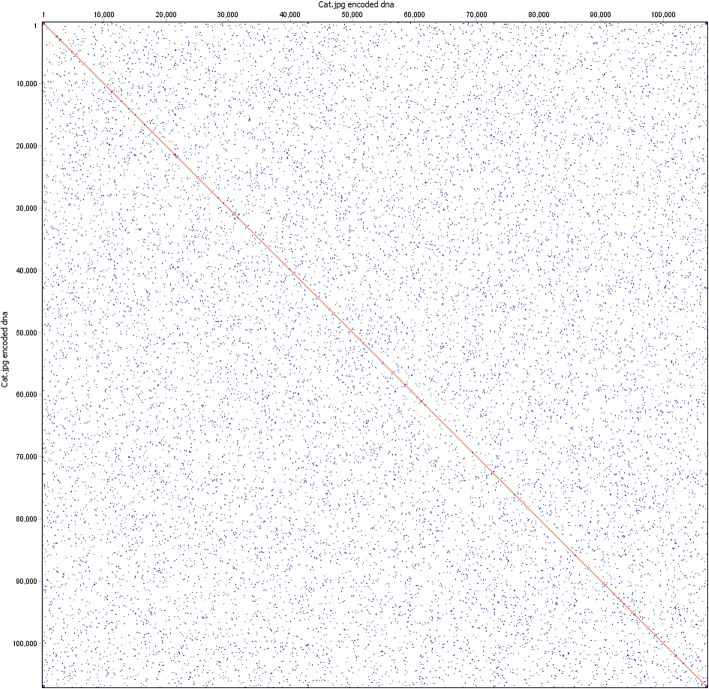
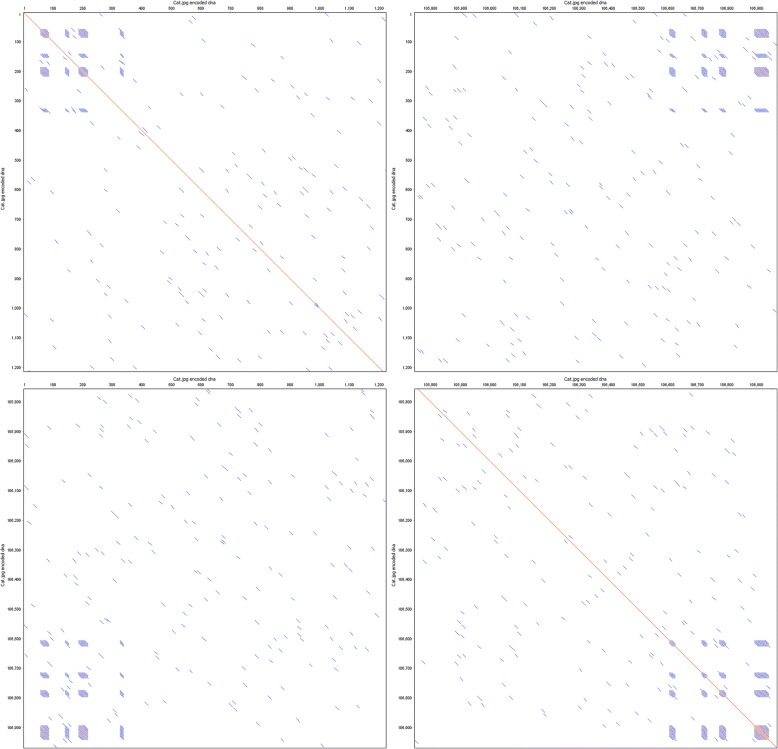
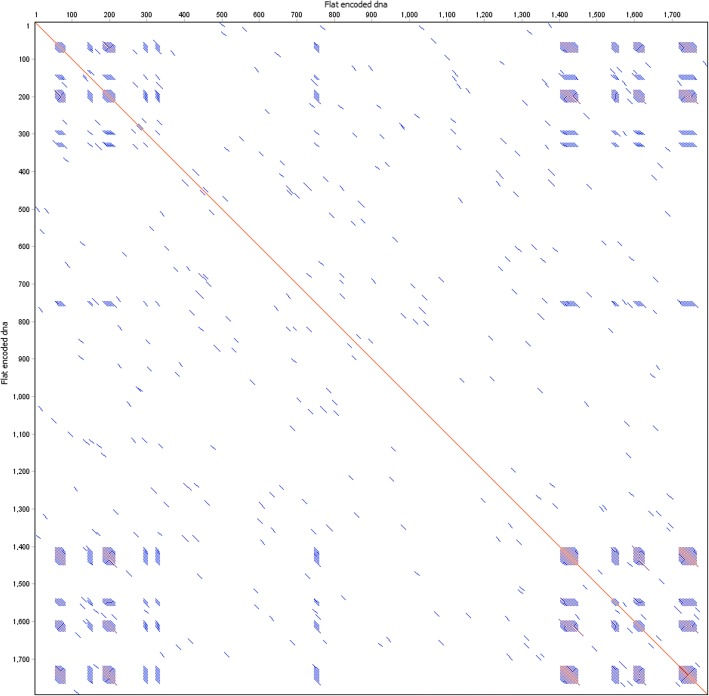
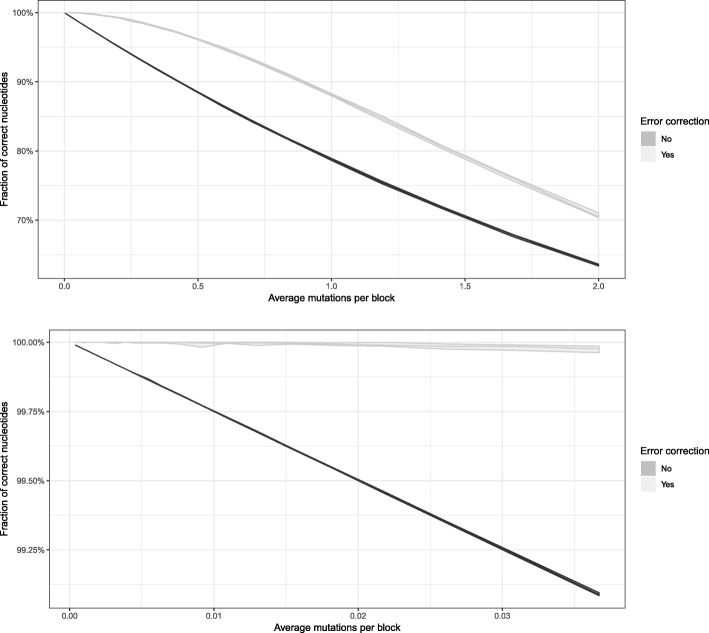
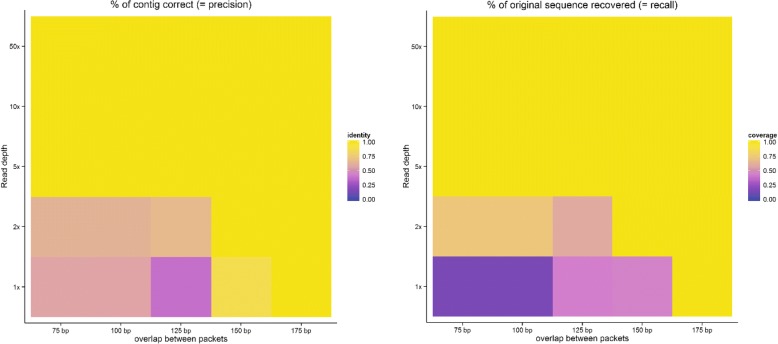
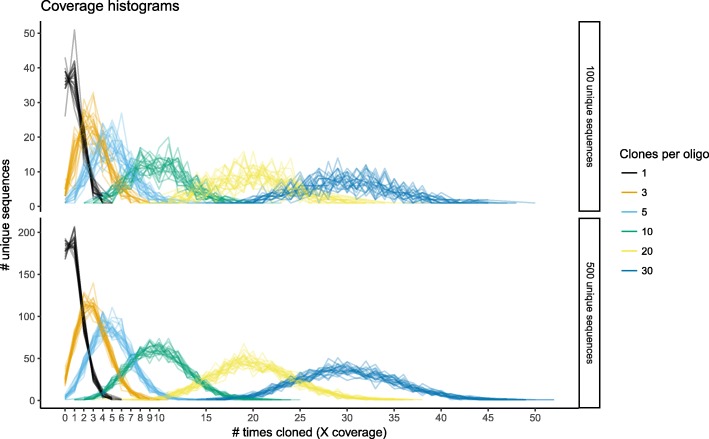
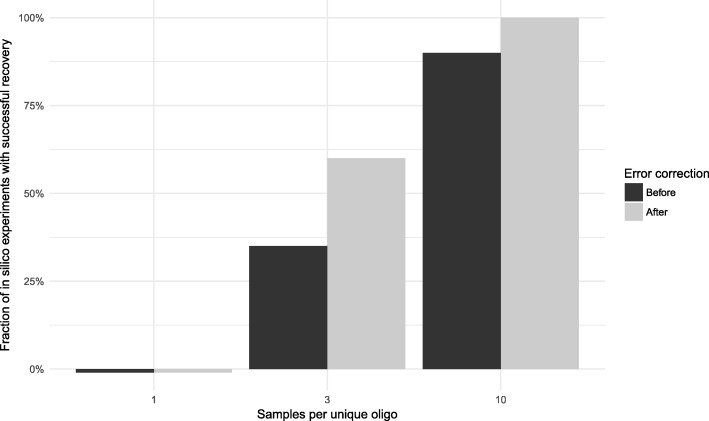
Results: To this end, we have developed and implemented an encoding scheme that is suitable for detecting and correcting errors that may arise during storage, writing, and reading, such as those arising from nucleotide substitutions, insertions, and deletions. We propose a scheme for parallelized long term storage of encoded sequences that relies on overlaps rather than the address blocks found in previously published work. Using computer simulations, we illustrate the encoding, sequencing, decoding, and recovery of encoded information, ultimately demonstrating the possibility of a successful round-trip read/write. These demonstrations show that in theory a precise control over error tolerance is possible. Even after simulated degradation of DNA, recovery of original data is possible owing to the error correction capabilities built into the encoding strategy. A secondary advantage of our method is that the statistical characteristics (such as repetitiveness and GC-composition) of encoded sequences can also be tailored without sacrificing the overall ability to store large amounts of data. Finally, the combination of the overlap-based partitioning of data with the LZMA compression that is integral to encoding means that the entire sequence must be present for successful decoding. This feature enables inordinately strong encryptions. As a potential application, an encrypted pathogen genome could be distributed and carried by cells without danger of being expressed, and could not even be read out in the absence of the entire DNA consortium.
Conclusions: We have developed a method for DNA encoding, using a significantly different fundamental approach from existing work, which often performs better than alternatives and allows for a great deal of freedom and flexibility of application.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures

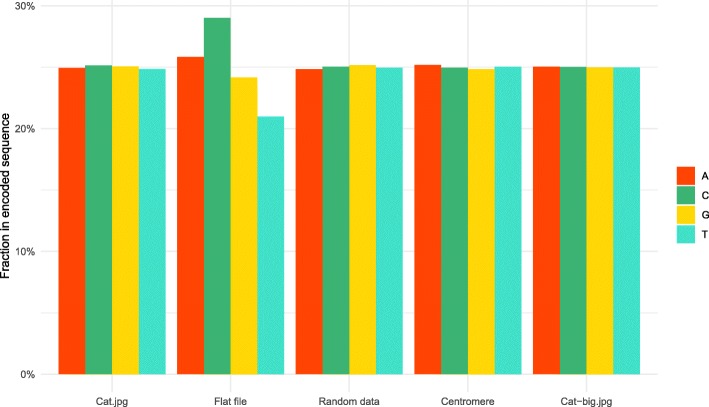
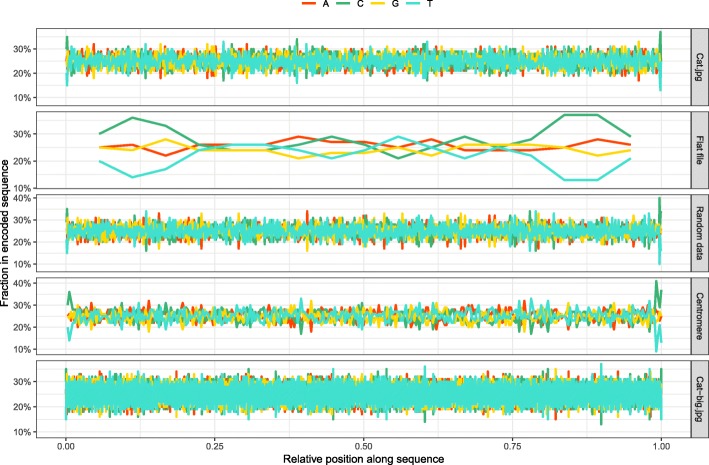
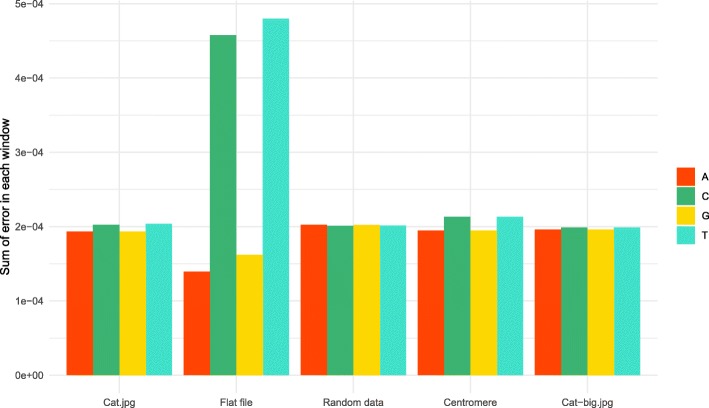
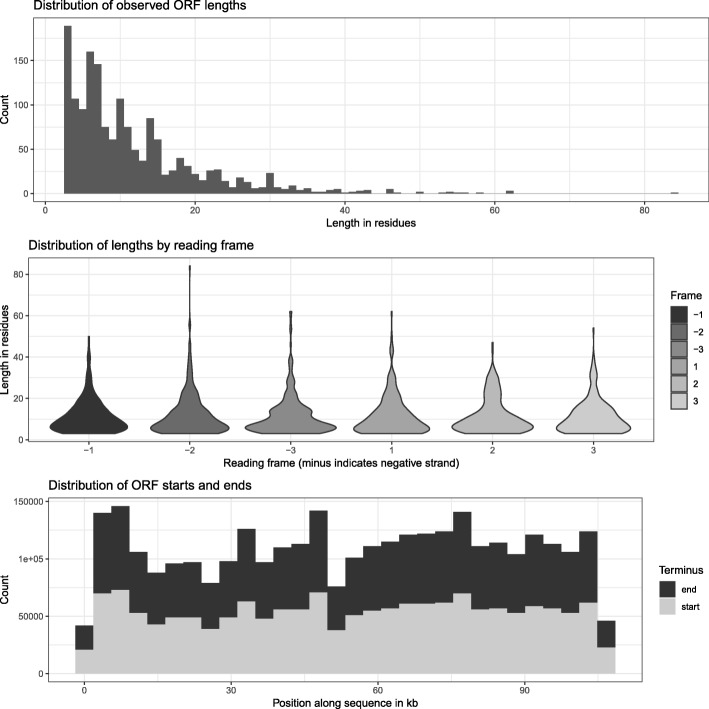

Similar articles
-
Orthogonal Information Encoding in Living Cells with High Error-Tolerance, Safety, and Fidelity.ACS Synth Biol. 2018 Mar 16;7(3):866-874. doi: 10.1021/acssynbio.7b00382. Epub 2018 Feb 21. ACS Synth Biol. 2018. PMID: 29429333
-
Next-generation digital information storage in DNA.Science. 2012 Sep 28;337(6102):1628. doi: 10.1126/science.1226355. Epub 2012 Aug 16. Science. 2012. PMID: 22903519
-
Random access in large-scale DNA data storage.Nat Biotechnol. 2018 Mar;36(3):242-248. doi: 10.1038/nbt.4079. Epub 2018 Feb 19. Nat Biotechnol. 2018. PMID: 29457795
-
Carbon-based archiving: current progress and future prospects of DNA-based data storage.Gigascience. 2019 Jun 1;8(6):giz075. doi: 10.1093/gigascience/giz075. Gigascience. 2019. PMID: 31220251 Free PMC article. Review.
-
Trends to store digital data in DNA: an overview.Mol Biol Rep. 2018 Oct;45(5):1479-1490. doi: 10.1007/s11033-018-4280-y. Epub 2018 Aug 2. Mol Biol Rep. 2018. PMID: 30073589 Review.
Cited by
-
Optimizing fountain codes for DNA data storage.Comput Struct Biotechnol J. 2024 Oct 26;23:3878-3896. doi: 10.1016/j.csbj.2024.10.038. eCollection 2024 Dec. Comput Struct Biotechnol J. 2024. PMID: 39559773 Free PMC article.
-
Robust direct digital-to-biological data storage in living cells.Nat Chem Biol. 2021 Mar;17(3):246-253. doi: 10.1038/s41589-020-00711-4. Epub 2021 Jan 11. Nat Chem Biol. 2021. PMID: 33432236 Free PMC article.
-
DNA storage: The future direction for medical cold data storage.Synth Syst Biotechnol. 2025 Mar 14;10(2):677-695. doi: 10.1016/j.synbio.2025.03.006. eCollection 2025 Jun. Synth Syst Biotechnol. 2025. PMID: 40235856 Free PMC article. Review.
-
DNA storage-from natural biology to synthetic biology.Comput Struct Biotechnol J. 2023 Feb 2;21:1227-1235. doi: 10.1016/j.csbj.2023.01.045. eCollection 2023. Comput Struct Biotechnol J. 2023. PMID: 36817961 Free PMC article. Review.
-
NOREC4DNA: using near-optimal rateless erasure codes for DNA storage.BMC Bioinformatics. 2021 Aug 17;22(1):406. doi: 10.1186/s12859-021-04318-x. BMC Bioinformatics. 2021. PMID: 34404355 Free PMC article.
References
-
- Bornholt J, Lopez R, Carmean DM, Ceze L, Seelig G, Strauss K. Proceedings of the Twenty-First International Conference on Architectural Support for Programming Languages and Operating Systems. 2016. A DNA-based archival storage system.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
